一种玉米RNA聚合酶III识别的启动子及其应用的制作方法
本发明涉及基因工程领域中一种玉米RNA聚合酶III识别的启动子及其应用。
背景技术:
RNA聚合酶是以一条DNA链或RNA链为模板催化核苷-5’-三磷酸生物合成RNA的酶,该类酶以模板为指导,以三磷酸核糖核苷为底物,通过磷酸二酯键聚合合成RNA。玉米等真核生物中,依据RNA聚合酶产物的不同,可以分为三类,分别为RNA聚合酶I、II和III。RNA聚合酶III(PolIII)主要负责转录snRNA,如5s rRNA等小分子RNA。PolIII可以在离启动子一个固定距离的位置开始转录合成RNA,遇到4-5个连续的U即终止,可以准确高效的产生小片段RNA。
启动子是一段位于结构基因5'端上游区的DNA序列,能活化RNA聚合酶,使之与模板DNA准确地相结合并具有转录起始的特异性。因为基因的特异性转录取决于酶与启动子能否有效地形成二元复合物,故RNA聚合酶如何有效地找到启动子并与之相结合是转录起始过程中首先要解决的问题。
CRISPR/Cas9是基于细菌或古细菌规律成簇的间隔短回文重复CRISPR(clustered regularly interspaced short palindromic repeats)介导的获得性免疫系统衍生而来的基因编辑技术,该技术通过RNA碱基互补配对识别DNA,指导Cas9蛋白切割识别的双链DNA,诱发同源重组(HDR,homologous directed repair)或非同源末端链接(NHEJ,non-homologous end-joining),进而实现目的DNA编辑。基于细菌II型免疫机制开发的基因编辑技术在植物,特别是作物遗传改良上具有重大的应用前景,该技术的基本要求之一就是受体细胞内表达单分子识别RNA(sgRNA,single guiding RNA),该分子负责识别特异性的基因编辑位点,然后介导结合Cas9蛋白行使DNA酶切活性,在设计的位点引入DNA双链断裂损伤,通过胞内的NHEJ或HDR修复途径引入突变。因此,sgRNA的表达是该技术的重要组成部分。
技术实现要素:
本发明所要解决的技术问题是提供一种玉米RNA聚合酶Ⅲ识别的启动子。
为解决上述技术问题,本发明首先提供了一种RNA聚合酶Ⅲ识别的启动子。
本发明所提供的一种RNA聚合酶Ⅲ识别的启动子,名称为ZmPolⅢca99,来源于玉米(Zea mays L.),是下述a)或b)或c)的DNA片段:
a)SEQ ID No.1所示的DNA分子;
b)与a)限定的DNA序列具有75%或75%以上同一性,且具有启动子功能的DNA分子;
c)在严格条件下与a)或b)限定的DNA序列杂交,且具有启动子功能的DNA分子。
上述启动子中,所述严格条件是在2×SSC,0.1%SDS的溶液中,在68℃下杂交并洗膜2次。每次5min 0.5×SSC,0.1%SDS的溶液中,在68℃下杂交并洗膜2次。每次15min。
上述75%或75%以上同一性,可为80%、85%、90%、95%以上的同一性。
本领域普通技术人员可以很容易地采用已知的方法,例如定向进化和点突变的方法,对本发明的启动子核苷酸序列进行突变。那些经过人工修饰的,具有与本发明分离得到的启动子核苷酸序列75%或者更高同一性的核苷酸,只要保持了表达靶基因的启动子活性,均是衍生于本发明的核苷酸序列并且等同于本发明的序列。
这里使用的术语“同一性”指与天然核酸序列的序列相似性。“同一性”包括与本发明的启动子核苷酸序列具有75%或更高,或85%或更高,或90%或更高,或95%或更高同一性的核苷酸序列。同一性可以用肉眼或计算机软件进行评价。使用计算机软件,两个或多个序列之间的同一性可以用百分比(%)表示,其可以用来评价相关序列之间的同一性。
为解决上述技术问题,本发明还提供了含有上述RNA聚合酶Ⅲ识别的启动子ZmPolⅢca99的生物材料。
本发明所提供的含有上述RNA聚合酶Ⅲ识别的启动子ZmPolⅢca99的生物材料,为下述B1)至B6)中至少一种:
B1)含有上述RNA聚合酶Ⅲ识别的启动子ZmPolⅢca99的表达盒;
B2)含有上述RNA聚合酶Ⅲ识别的启动子ZmPolⅢca99的重组载体、或含有B1)所述表达盒的重组载体;
B3)含有上述RNA聚合酶Ⅲ识别的启动子ZmPolⅢca99的重组微生物、或含有B1)所述表达盒的重组微生物、或含有B2)所述重组载体的重组微生物;
B4)含有上述RNA聚合酶Ⅲ识别的启动子ZmPolⅢca99的转基因植物细胞系、或含有B1)所述表达盒的转基因植物细胞系;
B5)含有上述RNA聚合酶Ⅲ识别的启动子ZmPolⅢca99的转基因植物组织、或含有B1)所述表达盒的转基因植物组织;
B6)含有上述RNA聚合酶Ⅲ识别的启动子ZmPolⅢca99的转基因植物器官、或含有B1)所述表达盒的转基因植物器官。
上述生物材料中,B2)所述重组载体可含有sgRNA编码基因,上述RNA聚合酶Ⅲ识别的启动子ZmPolⅢca99启动所述sgRNA编码基因的转录。
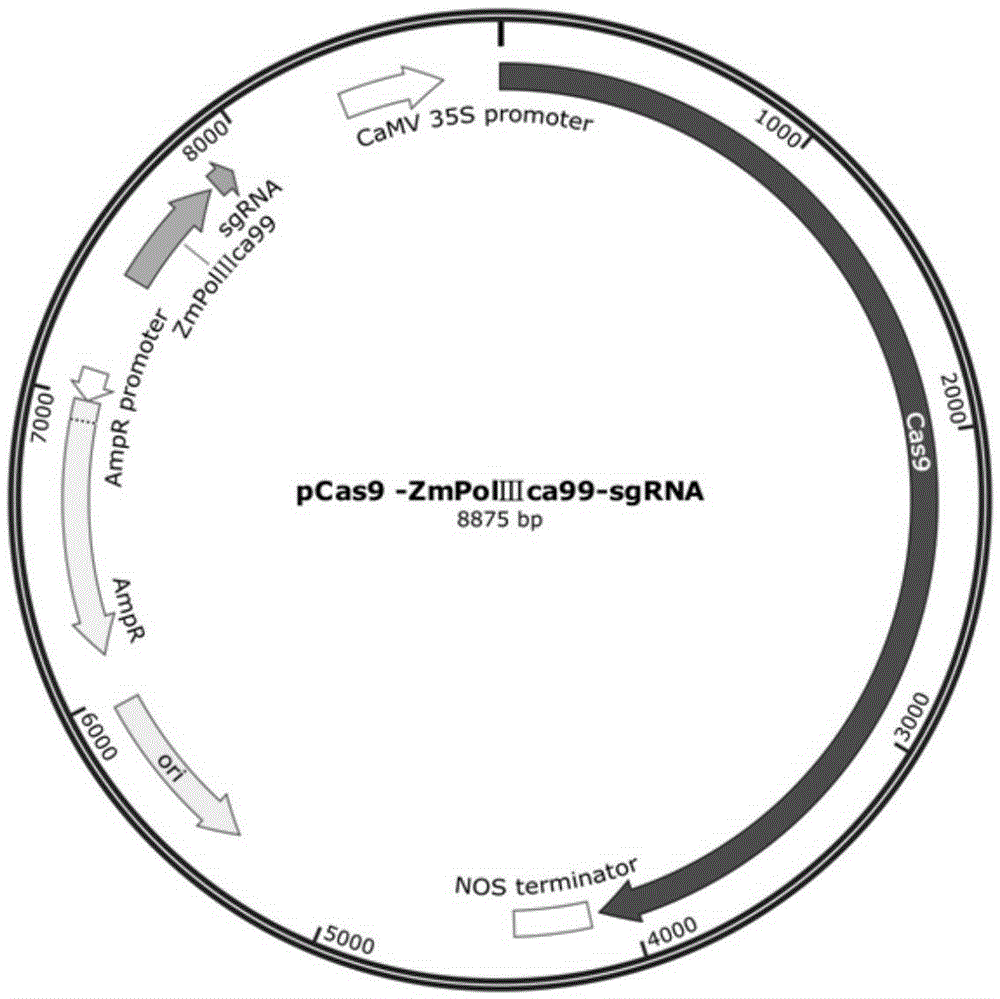
上述生物材料中,B2)所述重组载体具体可为重组载体pCas9-ZmPolⅢ ca99-sgRNA,其核苷酸序列为SEQ ID No.2。SEQ ID No.2的第7421-7816位所示的核苷酸序列为所述RNA聚合酶Ⅲ识别的启动子ZmPolⅢca99,SEQ ID No.2的第7817-7919位所示的核苷酸序列为sgRNA的编码基因,SEQ ID No.2的第7920-8875位所示的核苷酸序列为CaMV 35s启动子,SEQ ID No.2的第1-4116位所示的核苷酸序列为Cas9蛋白基因,SEQ ID No.2的第4139-4391位所示的核苷酸序列为NOS终止子,SEQ ID No.2的第5371-5959位所示的核苷酸序列为复制子ori,SEQ ID No.2的第6130-7095位所示的核苷酸序列为氨苄青霉素抗性基因。
上述与生物材料中,B3)所述重组微生物具体可为细菌,酵母,藻和真菌。其中,细菌可来自埃希氏菌属(Escherichia),欧文氏菌(Erwinia),根癌农杆菌属(Agrobacterium)、黄杆菌属(Flavobacterium),产碱菌属(Alcaligenes),假单胞菌属(Pseudomonas),芽胞杆菌属(Bacillus)等。
B4)所述的转基因植物细胞系,B5)所述的转基因植物组织和B6)所述的转基因植物器官不包括植物的繁殖材料。
上述重组载体pCas9-ZmPolⅢca99-sgRNA可通过使用PEG介导法、Ti质粒、Ri质粒、植物病毒载体、直接DNA转化、显微注射、电导、农杆菌介导、基因枪等常规生物学方法转化植物细胞或组织,并将转化的植物细胞或组织培育成植株。
为解决上述技术问题,本发明还提供了用于基因编辑的产品。
本发明所提供的用于基因编辑的产品,含有B2)所述重组载体,B2)所述重组载体可含有sgRNA编码基因,上述RNA聚合酶Ⅲ识别的启动子ZmPolⅢca99启动所述sgRNA编码基因的转录。
本发明所提供的上述RNA聚合酶Ⅲ识别的启动子ZmPolⅢca99在作为启动子中的应用也属于本发明保护的范围。
本发明所提供的上述RNA聚合酶Ⅲ识别的启动子ZmPolⅢca99在植物中启动sgRNA编码基因表达中的应用也属于本发明保护的范围。
为解决上述技术问题,本发明还提供了下述1)-12)中任一种应用:
1)所述RNA聚合酶Ⅲ识别的启动子ZmPolⅢca99在制备基因编辑载体中的应用;
2)所述RNA聚合酶Ⅲ识别的启动子ZmPolⅢca99在制备利用CRISPR/Cas9系统进行基因编辑的载体中的应用,所述载体中所述RNA聚合酶Ⅲ识别的启动子ZmPolⅢca99启动sgRNA编码基因的转录;
3)所述RNA聚合酶Ⅲ识别的启动子ZmPolⅢca99在制备基因编辑产品中的应用;
4)所述RNA聚合酶Ⅲ识别的启动子ZmPolⅢca99在植物基因编辑中的应用;
5)所述RNA聚合酶Ⅲ识别的启动子ZmPolⅢca99在培育转基因植物中的应用;
6)所述生物材料在制备基因编辑载体中的应用;
7)所述生物材料在制备利用CRISPR/Cas9系统进行基因编辑的载体中的应用,所述载体中所述RNA聚合酶Ⅲ识别的启动子ZmPolⅢca99启动sgRNA编码基因的转录;
8)所述生物材料在制备基因编辑产品中的应用;
9)所述生物材料在植物基因编辑中的应用;
10)所述生物材料在培育转基因植物中的应用;
11)所述产品在植物基因编辑中的应用;
12)所述产品在培育转基因植物中的应用。
上文中,所述植物可为单子叶植物或双子叶植物;所述单子叶植物具体可为玉米。
上文中,所述基因编辑可为基因删除、插入、替换等,具体可为基因删除。
实验证明,采用本发明启动子ZmPolⅢca99构建的重组载体pCas9-ZmPolⅢca99-sgRNA与载体TLR共转化玉米原生质体,对于目标序列进行了成功编辑,获得了三种Cas9蛋白剪切修复后的三种类型,即在同一个原生质体内只有eGFP蛋白,在同一个原生质体内只有DsRed2蛋白和在同一个原生质体内既有eGFP蛋白又有DsRed2蛋白的情况。在所检测的11120个实验组原生质体中发现488nm下发绿光原生质体数为106个,约占被检测原生质体总数的0.95%;563nm下发红光原生质体数为279个,约占被检测原生质体总数的2.51%;原生质体同时发出绿光和红光的共3个,约占被检测原生质体总数的0.03%;总突变原生质体数为388个,总突变率为3.49%。对照组1共检测7601个原生质体,488nm下发绿光原生质体数为7个,约占被检测原生质体总数的0.09%;563nm下发红光原生质体数为11个,约占被检测原生质体总数的0.14%;无同时发出绿光和红光的原生质体;总突变原生质体数为18个,总突变率为0.24%。对照组2共检测1667个原生质体,488nm下发绿光原生质体数为0个;563nm下发红光原生质体数为4个,约占被检测原生质体总数的0.24%;无同时发出绿光和红光的原生质体;总突变原生质体数为4个,总突变率为0.24%。对照组3共检测714个原生质体,488nm下发绿光原生质体数为0个;563nm下发红光原生质体数为2个,约占被检测原生质体总数的0.28%;无同时发出绿光和红光的原生质体;总突变原生质体数为2个,总突变率为0.28%。对照组4共检测1852个原生质体,488nm下发绿光原生质体数为0个,563nm下发红光原生质体数为5个,约占被检测原生质体总数的0.27%;原生质体同时发出绿光和红光的共0个;总突变原生质体数为5个,总突变率为0.27%。对照组5共检测10785个原生质体,488nm下发绿光原生质体数为84个,约占被检测原生质体总数的0.78%;563nm下发红光原生质体数为190个,约占被检测原生质体总数的1.76%;原生质体同时发出绿光和红光的共3个,约占被检测原生质体总数的0.03%;总突变原生质体数为277个,总突变率为2.57%。实验组的总突变率明显高于其它各个对照组,启动子ZmPolⅢca99是可以启动sgRNA编码基因转录的启 动子,是RNA聚合酶III识别的启动子(RNA聚合酶III启动子),能够参与CRISPR/Cas9系统的基因组编辑,在植物育种中具有重要的意义。
附图说明
图1为载体PN11-eGFP的结构示意图。
图2为重组载体pCas9-ZmPolⅢca99-sgRNA的结构示意图。
图3为载体The Traffic Light Reporter(TLR)的结构示意图。
图4为玉米自交系-郑58的原生质体活性检测图。其中,A为488nm波长下观察到的活性原生质体荧光图;B为白光下观察到的玉米原生质体图像。
图5为重组载体pCas9-ZmPolⅢca99-sgRNA的功能验证流程图。其中,Binary vector 1代表载体TLR;Binary vector 2代表重组载体pCas9-ZmPolⅢca99-sgRNA。
图6为重组载体pCas9-ZmPolⅢca99-sgRNA与载体TLR共转化玉米原生质体的共聚焦显微镜检测突变图。其中,A为同一个原生质体既含有eGFP蛋白又有DsRed2蛋白的荧光突变图,B为同一个原生质体只含有eGFP蛋白的荧光突变图,C为同一个原生质体只含有DsRed2蛋白的荧光突变图,D为A对应的样品在白光下观察的结果,E为B对应的样品在白光下观察的结果,F为C对应的样品在白光下观察的结果。
图7为各组玉米原生质体的流式细胞仪检测结果图。其中,A中横轴FSC表示前向散射光,该数值与细胞大小相关,纵轴SSC表示侧向散射光,该数值与细胞内结构复杂程度相关,象限内所有点表示所有原生质体数目,P1区是依据FSC与SSC值在记录的原生质体中选择原生质体完整度较好的群体并进行统计;B中对照组1为玉米自交系郑58的原生质体的流式细胞仪统计结果,对照组2为转入重组载体pCas9-ZmPolⅢca99-sgRNA的原生质体的流式细胞仪统计结果,对照组3为转入载体TLR的原生质体的流式细胞仪统计结果,对照组4为转入重组载体pCas9-sgRNA与载体TLR的原生质体的流式细胞仪统计结果,对照组5为转入重组载体pCas9-U6-sgRNA与载体TLR的原生质体的流式细胞仪统计结果,实验组为重组载体pCas9-ZmPolⅢca99-sgRNA与载体TLR共转化玉米原生质体的流式细胞仪统计结果;B中横坐标为488nm检测绿光信号,纵坐标为563nm检测红光信号,Q1象限表示只发红光的原生质体个数,Q2象限为在一个原生质体中同时检测到绿光和红光两种信号,Q3象限表示没有萤光信号原生质体个数,Q4象限表示只发绿光的原生质体个数。
具体实施方式
下面结合具体实施方式对本发明进行进一步的详细描述,给出的实施例仅为了阐明本发明,而不是为了限制本发明的范围。
下述实施例中的实验方法,如无特殊说明,均为常规方法。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
下述实施例中的玉米自交系-郑58,为河南农业科学院育成品种郑单958的母本,为国内玉米育种上常用材料,本材料取自中国农业科学院作物科学研究所国家种质资源库。
下述实施例中的pEASY-Blunt Cloning Kit、Trans5α化学感受态细胞、pEASY-Uni Seamless Cloning and Assembly Kit均为北京全式金生物技术有限公司的产品;纤维素酶R-10和离析酶R-10均为Japan Yakult Honsha,Tokyo公司的产品。
下述实施例中的二乙酸荧光素(FDA)为国药集团化学试剂有限公司的产品。
实施例1、含有玉米RNA聚合酶Ⅲ识别的启动子ZmPolⅢca99(下文简称启动子ZmPolⅢca99)的CRISPR/Cas9基因编辑载体的构建及其功能验证
一、启动子ZmPolⅢca99的克隆
取玉米自交系-郑58种子3-4粒,消毒浸种后,将其放入潮湿的石英沙盘中培育,待长出叶片,提取叶片基因组备用。以叶片基因组为模板,利用引物对ZmPolⅢca99-F(5’-AATTGGCCCTTACAAAATAG-3’)与ZmPolⅢca99-R(5’-GGAGCGGTGGTC GCAGCTG-3’)进行PCR扩增,得到PCR扩增片段。将所得PCR扩增片段利用pEASY-Blunt Cloning Kit进行亚克隆,实验操作参照说明述进行,得到重组载体1。将获得的重组载体1转化入Trans5α化学感受态细胞中,培养过夜,挑取单克隆,扩大培养并测序,结果表明重组载体1中含有SEQ ID No.1所示的启动子ZmPolⅢca99序列,将该重组载体1命名为ZmPolⅢca99亚克隆载体。
二、含有启动子ZmPolⅢca99的CRISPR/Cas9基因编辑载体的构建
含有启动子ZmPolⅢca99的CRISPR/Cas9基因编辑载体,其核苷酸序列为SEQ ID No.2。SEQ ID No.2的第7421-7816位所示的核苷酸序列为所述RNA聚合酶Ⅲ识别的启动子ZmPolⅢca99,SEQ ID No.2的第7817-7919位所示的核苷酸序列为sgRNA的编码基因,SEQ ID No.2的第7920-8875位所示的核苷酸序列为CaMV 35s启动子,SEQ ID No.2的第1-4116位所示的核苷酸序列为Cas9蛋白基因,SEQ ID No.2的第4139-4391位所示的核苷酸序列为NOS终止子,SEQ ID No.2的第5371-5959位所示的核苷酸序列为复制子ori,SEQ ID No.2的第6130-7095位所示的核苷酸序列为氨苄青霉素抗性基因。具体制备方法如下:
采用限制性内切酶SalI和XbaI双酶切载体PN11-eGFP(图1),获得线性载体1。利用引物对Cas9(SalI)-F(5’-caccatcaccatcacgccatggacaagaagtacagc-3’)与Cas9(XbaI)-R(5’-ttcgagctctatcgatcaatcaggatcgtcgcctcccagc-3’)从载体CPB中分离并扩增携带NLS的Cas9蛋白基因片段,并在携带NLS的Cas9蛋白基因片段上游增加与线性化载体的SalI酶切位点端同源的15bp核苷酸片段,下游增加与线性载体1的XbaI端同源的15bp核苷酸片段,得到Cas9蛋白基因片段1。通过pEASY-Uni Seamless Cloning and Assembly Kit将线性载体1与Cas9蛋白基因片段1实现无缝连接克隆,得到重组载体2。将重组载体2转化入Trans5α化学感受态细胞中,培养过夜,挑取单克隆,扩大培养并测序。测序结果表明重组载体2中含有SEQ ID No.2的第1-4116位所示的核苷酸序列,为Cas9蛋白基因(Cas9蛋白的编码区),将该重组载体2命名为重组载体pCaMV 35s::cas9::NOS。
通过限制性内切酶HindIII酶切重组载体pCaMV 35s::cas9::NOS,生成线性化载体2。利用引物对ZmPolⅢca99(HindIII)-F与ZmPolⅢca99(Seed)-R从ZmPolⅢca99亚克隆载体中扩增启动子ZmPolⅢca99片段,并使启动子ZmPolⅢca99片段上游携带与线性化载体2一端具有15bp同源序列的核苷酸片段,启动子ZmPolⅢca99片段下游携带Cas9目标序列中的GTGATCGCGGCCGTGTATGC片段,得到启动子ZmPolⅢca99片段1。通过PCR技术利用引物对sgRNA(Seed)-F与sgRNA(HindIII)-R以sgRNA scaffold基因为模板,在sgRNA scaffold基因上游加GTGATCGCGGCCGTGTATGC序列,下游增加与线性化载体2具有15bp同源序列的核苷酸片段,得到sgRNA的编码基因片段1。通过pEASY-Uni Seamless Cloning and Assembly Kit将线性载体2、启动子ZmPolⅢca99片段1和sgRNA的编码基因片段1实现无缝连接克隆,得到重组载体3。将重组载体3转化入Trans5α化学感受态细胞中,培养过夜,挑取单克隆,扩大培养并测序。测序结果表明重组载体3的核苷酸序列为SEQ ID No.2,将该重组载体3命名为重组载体pCas9-ZmPolⅢca99-sgRNA,即为含有启动子ZmPolⅢca99的CRISPR/Cas9基因编辑载体(图2)。
SEQ ID No.2的第7421-7816位的核苷酸序列为所述RNA聚合酶Ⅲ识别的启动子ZmPolⅢca99。
将SEQ ID No.2中的第7421-7816位所示的启动子ZmPolⅢca99替换为玉米U6启动子(核苷酸序列为SEQ ID No.4),保持SEQ ID No.2的其它核苷酸序列不变,得到对照载体,将其命名为重组载体pCas9-U6-sgRNA。
将SEQ ID No.2中的第7421-7816位所示的ZmPolⅢca99去除,保持SEQ ID No.2的其它核苷酸序列不变,得到对照载体,将其命名为重组载体pCas9-sgRNA。重组载体pCas9-sgRNA与重组载体pCas9-ZmPolⅢca99-sgRNA的区别仅在于重组载体pCas9-sgRNA不含ZmPolⅢca99。
所用引物序列如下:
sgRNA(Seed)-F:GTGATCGCGGCCGTGTATGCGTTTTAGAGCTAGAAATAGCA;
sgRNA(HindIII)-R:TGCAGGCATGCAAGCTTAAAAAAAGCACCGACTCGGTGC;
ZmPolⅢca99(HindIII)-F:GGCCAGTGCCAAGCTTTTGTATTTTTGTTTCTTGGC;
ZmPolⅢca99(Seed)-R:GCATACACGGCCGCGATCACGGAGTGGTAGTCGCAGCTG。
三、表达载体The Traffic Light Reporter(简称为载体TLR)的构建
载体TLR的核苷酸序列为SEQ ID No.3所示,SEQ ID No.3的第279-1204位所示的核苷酸序列为CaMV 35s启动子,SEQ ID No.3的第1260-1279位所示的核苷酸序列为sgRNA介导的Cas9蛋白的目标序列,SEQ ID No.3的第1280-1282位所示的核苷酸序列为PAM位点,SEQ ID No.3的第1282-1995位所示的核苷酸序列为eGFP蛋白基因,SEQ ID No.3的第1998-2675位所示的核苷酸序列为DsRed2蛋白基因(DsRed-2蛋白基因),SEQ ID No.3的第2762-3014位所示的核苷酸序列为NOS终止子,SEQ ID No.3的第3994-4582位所示的核苷酸序列为复制子ori,SEQ ID No.3的第4753-5718位所示的核苷酸序列为氨苄青霉素抗性基因,SEQ ID No.3的第1996-1997位所示的核苷酸序列为两个胞嘧啶脱氧核苷酸使得DsRed2蛋白基因也发生了生移码突变。Cas9蛋白的目标序列为5’-TGTGATCGCGGCCGTGTATGCGG-3’。具体制备方法如下:
采用限制性内切酶SalI和XbaI双酶切载体PN11-eGFP(图1),获得线性载体1。利用引物对GFP(Seed)-F与GFP(DsRed)-R从载体PN11-eGFP中分离并扩增eGFP蛋白基因,并将Cas9蛋白目标序列增加到eGFP蛋白基因序列上游ATG下游,并且在eGFP蛋白基因的3’末端去掉终止子,替换成两个胞嘧啶核糖核苷酸,得到片段1。利用引物对GFP(PN11-salI)-F与GFP(DsRed)-R在片段1中的Cas9蛋白目标序列上游增加与线性化载体1的限制性内切酶SalI酶切位点端同源的15bp核苷酸片段,得到片段2。利用引物对DsRed(GFP)-F与DsRed(PN11)-R从载体pDsRed2中分离DsRed2蛋白基因,并且在DsRed2蛋白基因序列3’末端增加与线性载体1的限制性内切酶XbaI酶切位端同源的15bp核苷酸片段,得到片段3。通过pEASY-Uni Seamless Cloning and Assembly Kit将线性载体1、片段2与片段3实现无缝连接克隆,得到重组载体4。将重组载体4转化入Trans5α化学感受态细胞中,培养过夜,挑取单克隆,扩大培养并测序。测序结果表明重组载体4的核苷酸序列为SEQ ID No.3,将该重组载体4命名为载体TLR(图3)。
所用引物序列如下:
GFP(PN11-salI)-F:TCACGCCATGGTCGACGTACTAGTGATGTGTGATCGCGGCCGTGTATGCGGGTG
GFP(Seed)-F:GCGGCCGTGTATGCGGGTGAGCAAGGGCGAGGAGCTG
GFP(DsRed)-R:gacgttctcggaggaggccatGGCTTGTACAGCTCGTCC
DsRed(GFP)-F:atggcctcctccgagaacgtc
DsRed(PN11)-R:GGTGATGCATTCCCGGGTCTAGACCCTTTctacaggaacaggtggtg
四、含有启动子ZmPolⅢca99的CRISPR/Cas9基因编辑载体的功能验证
1、玉米原生质体的制备
1)玉米的培养
将玉米自交系-郑58玉米种子浸泡于30%过氧化氢中5min,蒸馏水清洗3遍,75%酒精消毒1h,蒸馏水清洗3遍。将消完毒的种子浸泡于无菌水中6-8h,沥干水分将种子置于潮湿纱布上30℃孵育2天,再将发好芽的玉米放置在盛有石英砂的苗盘中,室温黑暗下培养约1周,直至到达玉米2叶期。
2)玉米原生质体的分离
将步骤1)中的玉米叶片去除叶片两端部分和主叶脉后称取1g,切成宽度为的0.5mm的细丝,把切好的叶片移入含有15mL纤维素酶酶解液(纤维素酶R100.225g,离析酶R100.075g,D-甘露醇1.64g,浓度为0.3M的KCl溶液1mL,浓度为0.3M(pH 5.7)的MES(2-(N-吗啉)乙烷磺酸)溶液1mL,去离子水10mL,55℃水浴锅中加热10min,冷却到室温后,加入浓度为0.15M的CaCl2溶液1mL,浓度为75mM的β-巯基乙醇溶液1mL,浓度为1.5%BSA溶液1mL,使用0.45μm滤膜过滤后使用)的三角瓶中,28℃黑暗中真空抽吸30min,放置黑暗下消化3h,然后转入28℃恒温摇床在60r/min条件下摇1h,得到玉米原生质体的酶解液。
3)原生质体纯化
用等量的提前遇冷W5溶液(154mmol/L NaCl;125mmol/L CaCl2;5mmol/L KCl;2mmol/L MES;pH 5.7)稀释步骤2)获得的玉米原生质体的酶解液,得到稀释后的玉米原生质体的酶解液。先用W5溶液润湿200目筛子,将稀释后的玉米原生质体的酶解液采用W5溶液润湿的200目筛子过滤,将滤液收集于50mL圆底离心管中,在70g条件下离心3min,弃上清液,再次向沉淀中加入10mL的W5溶液重悬,然后置于黑暗下冰浴30min,将冰浴完的样品在70g条件下离心10min,离心后充分吸除上清液,向沉淀中加入适量的MMg溶液(4mmol/L MES,0.6mol/L甘露醇,15mmol/L MgCl2),得到纯化后的原生质体溶液。
4)原生质体计数与活力检测
用浓度为0.01%的二乙酸荧光素(FDA)溶液染色,FDA本身无荧光,可以自由的通过细胞膜进入细胞质,当FDA在细胞质内非特异性的脂质酶催化下,FDA生成不可穿过细胞膜的荧光素,这种荧光素在480nm的激发下产生530nm左右的绿色荧光。死细胞由于细胞膜不完整,产生的荧光素很快扩散到溶液中,所以不显荧光;然而荧光素不可穿过活体细胞膜,所以活细胞显示绿色荧光。用血球计数板在荧光显微镜下统计发绿色荧光的原生质体数和原生质体总数,重复取样3次,每个样品计数3次。
原生质体活力=(发绿色荧光的原生质体数/原生质体总数)×100%;
经FDA染色后,玉米自交系-郑58原生质体活性均在95%以上,图4为玉米自交系-郑58的原生质体活性检测图。
2、原生质体转化
将步骤二构建的重组载体pCas9-ZmPolⅢca99-sgRNA与步骤三构建的载体TLR按照1:1质量比混合,得到载体混合物1;利用MMg溶液将玉米自交系-郑58的原生质体的终浓度调节为1×106个/mL,原生质体转化采用PEG-Ca2+介导法进行。将100μL原生质体溶液中加入20μg载体混合物1,混匀后加入110μL的45%的PEG-Ca2+溶液(10mL45%的PEG4000转化试剂:PEG40004.5g,去离子水2mL,浓度为0.8M的甘露醇溶液1mL,浓度为1M的氯化钙溶液1mL,现用现配),混匀后于23℃黑暗下孵育20min,然后加入440μL W5溶液终止反应,得到反应液。将反应液在70g条件下离心30min,吸去上清,向沉淀中加入260μL W5溶液,混匀后加入24孔细胞培养板中,加入5%胎牛血清冲洗10s,在23℃培养12-16h,得到转入重组载体pCas9-ZmPolⅢca99-sgRNA与载体TLR的原生质体,记为实验组。
同时设置四个对照原生质体:对照组1为玉米自交系-郑58的原生质体,对照组2为转入重组载体pCas9-ZmPolⅢca99-sgRNA的原生质体,对照组3为转入载体TLR的原生质体,对照组4为转入重组载体pCas9-sgRNA与载体TLR的原生质体,对照组5为转入重组载体pCas9-U6-sgRNA与载体TLR的原生质体,这些原生质体的制备方法同实验组。
3、显微镜观察与检测计数
将转化过夜的原生质体装入2mL圆底离心管中,在70g条件下离心3min,收集沉淀,加入适量W5溶液后,吸取少量原生质体悬液置于载玻片中央,盖上盖玻片,在Zeiss LSM700激光共聚焦显微镜下观察DsRed2蛋白与eGFP蛋白的表达情况。
将转化过夜的原生质体装入2mL离心管中,在70g条件下离心3min,加入适量W5溶液后,放入上样管中,在BD FACSAriaIII流式细胞分选仪上进行检测计数。
基于原生质体CRISPR/Cas9技术,利用启动子ZmPolⅢca99所产生的20nt sgRNA引导CRISPR/Cas9核酸酶,特异地靶向载体TLR(binary vector1)(图5)中报告基因eGFP蛋白基因与DsRed2蛋白基因上游一个特定的目标序列23bp区间中的20bp,引起DNA双链损伤,并引入短的插入或缺失突变(Indel,insertion and deletion)。由于eGFP蛋白基因的起始密码子ATG与eGFP第二个密码子GTG之间插入了CRISPR/Cas9系统的靶标序列(5’-TGTGATCGCGGCCGTGTATGCGG-3’),所以导致eGFP蛋白基因发生移码突变,eGFP蛋白基因不能正常翻译表达产生eGFP蛋白,而在eGFP蛋白基因后面增加两个胞嘧啶脱氧核苷酸使得DsRed2蛋白基因也发生了生移码突变,DsRed2蛋白基因不能正常翻译表达产生DsRed2蛋白,所以当靶标序列区域被CRISPR/Cas9切割后发生修复时,当缺失3n+2bp或插入3n-1bp个核苷酸时,则eGFP蛋白基因开放阅读框(ORF,open reading frame)被恢复,表达有功能的蛋白,在 488nm下发绿光;当缺失3n+1或插入3n-2bp个核苷酸时,则DsRed2蛋白基因ORF被恢复,在563nm下发红光。因此,通过本系统可以统计产生不同类型突变的频率,即分析目标序列下游报告基因移码突变的频率。含有启动子ZmPolⅢca99的重组载体pCas9-ZmPolⅢ-sgRNA ca99(binary vector 2)(图5)和含有报告基因eGFP蛋白基因与DsRed2蛋白基因的载体TLR(binary vector 1)共转化玉米原生质体通过上述突变的频率来评估启动子ZmPolⅢca99在CRISPR/Cas9系统中的效率。
共聚焦显微镜结果显示(图6)实验组的原生质体获得了三种Cas9蛋白剪切修复后的三种类型,即在同一个原生质体内只有eGFP蛋白(绿光),在同一个原生质体内只有DsRed2蛋白(红光)和在同一个原生质体内既有eGFP蛋白又有DsRed2蛋白的情况。
流式细胞仪计数结果表明(图7和表1):在所检测的11120个实验组原生质体中发现488nm下发绿光原生质体数为106个,约占被检测原生质体总数的0.95%;563nm下发红光原生质体数为279个,约占被检测原生质体总数的2.51%;原生质体同时发出绿光和红光的共3个,约占被检测原生质体总数的0.03%;总突变原生质体数为388个,总突变率为3.49%。对照组1共检测7601个原生质体,488nm下发绿光原生质体数为7个,约占被检测原生质体总数的0.09%;563nm下发红光原生质体数为11个,约占被检测原生质体总数的0.14%;无同时发出绿光和红光的原生质体;总突变原生质体数为18个,总突变率为0.24%。对照组2共检测1667个原生质体,488nm下发绿光原生质体数为0个;563nm下发红光原生质体数为4个,约占被检测原生质体总数的0.24%;无同时发出绿光和红光的原生质体;总突变原生质体数为4个,总突变率为0.24%。对照组3共检测714个原生质体,488nm下发绿光原生质体数为0个;563nm下发红光原生质体数为2个,约占被检测原生质体总数的0.28%;无同时发出绿光和红光的原生质体;总突变原生质体数为2个,总突变率为0.28%。对照组4共检测1852个原生质体,488nm下发绿光原生质体数为0个,563nm下发红光原生质体数为5个,约占被检测原生质体总数的0.27%;原生质体同时发出绿光和红光的共0个;总突变原生质体数为5个,总突变率为0.27%。对照组5共检测10785个原生质体,488nm下发绿光原生质体数为84个,约占被检测原生质体总数的0.78%;563nm下发红光原生质体数为190个,约占被检测原生质体总数的1.76%;原生质体同时发出绿光和红光的共3个,约占被检测原生质体总数的0.03%;总突变原生质体数为277个,总突变率为2.57%。实验组的总突变率明显高于其它各个对照组,证明启动子ZmPolⅢca99是可以启动sgRNA转录的启动子,是RNA聚合酶III识别的启动子(RNA聚合酶III启动子)。
总突变原生质体数=发绿光原生质体数+发红光原生质体数+同时发绿光和红光原 生质体数;
总突变率=总突变原生质体数/原生质体总数×100%。
表1、各组原生质体的流式细胞仪检测结果
- 还没有人留言评论。精彩留言会获得点赞!