制备碳酸二芳基酯的方法与流程
由单酚和光气制备纯碳酸二芳基酯的方法是已知的。碳酸二芳基酯(例如碳酸二苯酯,“DPC”)的制备通常借助连续法——通过制备光气和随后使单酚和光气在惰性溶剂中在碱和氮催化剂存在下在界面中反应来进行。
原则上在文献中描述了通过例如相界面法制备碳酸二芳基酯,参见例如Chemistry and Physics of Polycarbonates, Polymer Reviews, H. Schnell, 第9卷, John Wiley and Sons, Inc. (1964), 第50/51页。
在此根据相界面法,溶解在溶剂和水中的反应物互相反应。这些方法的缺点是通过蒸馏从溶剂中分离碳酸二芳基酯并将其重新后处理,以及作为废产物的含氯化钠的水相,对此只有有限的使用可能性并且任选需要非常复杂的后处理步骤。
因此,已经开发出单酚的所谓的直接光气化的方法,其中反应物光气和单酚不在相界面法中在碱金属氢氧化物溶液存在下反应,而是在熔体中在催化剂存在下反应,优选不使用另外的溶剂,以形成碳酸二芳基酯和氯化氢而非氯化钠。
因此,例如,US 2,362,865 (A)描述了通过在170℃至250℃的温度下使用Al-或Ti-酚盐的单酚直接光气化来制备碳酸二芳基酯的方法,但其中没有描述催化剂的再循环,更没有描述分离方法。
EP 2 371 806 A和EP 2 371 807 A都同样描述了通过在20℃至240℃的温度下使用金属卤化物或金属酚盐的单酚直接光气化来制备碳酸二芳基酯的方法。同样没有描述催化剂再循环到该方法中。
EP 1 234 845 A同样描述了单酚在120℃至190℃的温度下在熔体中与特别纯的光气的反应。使用基于所用单酚计0.1至10摩尔%的量的含氮化合物,例如吡啶作为催化剂。此公开也没有给出催化剂再循环到该方法中的指示。吡啶与氯化氢形成难挥发的盐(沸点:222-224℃),其不容易蒸馏出。根据EP 1 234 845 A的教导,因此首先用氢氧化钠中和反应混合物,以可蒸馏出水、游离吡啶和过量酚的混合物。
WO 2011007001描述了从反应混合物中凝华吡啶盐酸盐。为此,在全油泵真空中将二氯硅烷-吡啶加合物加热到200℃,由此使吡啶盐酸盐挥发并作为固体沉淀。
此外,在许多其它专利,如WO 2008/114750 A1、JP 2008-231073 A、JP 2009-114195 A、JP 09-278714 A、JP 09-100256 A、JP 10-245366 A、JP 11-012230 A中描述了单酚在熔体中在均匀可溶的含氮催化剂的存在下与光气反应以形成碳酸二芳基酯。
JP 10-077250 A、JP 09-24278 A和EP 1 234 845 A提到催化剂的可能的再循环,但没有提到具体从产物中分离催化剂和着眼于再循环的催化剂后处理方法。此外,提到在反应混合物的中和和洗涤过程中引入水溶液,特别是水和/或氢氧化钠溶液。
US 5 239 106教导了通过与苯酚的1:1加合物的结晶从含催化剂的反应溶液中分离碳酸二苯酯。但是,在此没有描述催化剂的分离和再循环。
这些公开无一提供将催化剂,例如吡啶再循环到该方法中的方法的令人满意的指示。特别地,在使用碱性水溶液的中和步骤后经水相分离出催化剂。
特别地,现有技术没有给出其中没有如上文引用的文献中那样借助含水添加剂中和盐酸盐(这受制于上述缺点)的从含产物的料流中分离催化剂的方法的具体实例。
这些公开无一描述用于制备碳酸二芳基酯的完全无水和无废水的方法。
现有技术中已知的方法因此不足以在催化剂再循环方面满足经济和生态的高要求并另外确保产物(其本身是进一步化学工艺的原材料)的高纯度。
但是,工业方法必须考虑经济方面。在此,催化剂的再循环属于在这点上评估的重要方面,因为催化剂的高度或甚至完全的排放意味着经济劣势并造成不合意的环境污染。形成的废水必须用极高费用净化,这对水处理厂带来巨大的挑战。在现有技术的方法中,要么需要高技术支出才能实现催化剂的再循环,要么安排催化剂的部分或完全排放。在这两种情况下都出现额外的废水流。
在直接光气化法中,提供催化剂的有效再循环是最重要的。此外,不仅在反应过程中,在后处理中也应该尽可能避免使用水溶液。这是因为含有机物质的废水首先必须以复杂的方式净化,然后再弃置。
因此,技术目的是开发省略附加原材料如氢氧化钠和水的根据单酚在熔体中的直接光气化法制备碳酸二芳基酯的方法,所述方法通过减少料流排放(清除物(Purge))而经济地运行并提供最终产物的恒定良好品质。
现在令人惊讶地发现,当使用任选取代的吡啶或其盐作为催化剂时,形成的盐酸盐、氯化氢和碳酸二苯酯可以通过蒸馏将彼此分离并同时可以获得高纯度的碳酸二芳基酯。令人惊讶地,在该蒸馏过程中没有发生吡啶盐酸盐的凝华,因为该盐保持溶解在分离出的低沸物相中并甚至在室温下也与酚形成液体混合物。
优选省略反应溶液或母液的中和和/或水的添加。这带来特别经济和生态的方法方式。
本发明因此提供一种通过在游离形式和/或其盐酸盐形式的至少一种任选取代的吡啶作为催化剂的存在下使至少一种单酚与光气和/或至少一种氯甲酸芳基酯反应来制备碳酸二芳基酯,优选碳酸二苯酯的方法,其中
a) 所述反应在反应器中在1-50巴(绝对)的压力下进行,
b) 将反应混合物从反应器转移到单级或多级蒸馏装置中,
c) 在至少一个蒸馏塔的顶部分离出含催化剂的馏出物,
d) 将所述含催化剂的馏出物至少部分再循环到步骤a)的反应器中,
e) 经塔的侧流分离出碳酸二芳基酯并任选进一步提纯。
在步骤a)至e)中都优选不使用水溶液。
步骤a)中的反应优选在高于80℃的温度下进行,以避免所得碳酸二芳基酯以固体形式沉淀。反应物的反应可以在常压或略微减小的压力下,也可以在最多50巴(绝对)的升高的压力下进行。在此,根据工艺条件,光气可以存在于凝聚相中或溶解在液相中。通过这种方法制成的碳酸二芳基酯由于它们的高纯度而特别适用于根据熔融酯交换法由碳酸二芳基酯和双酚制备高纯度的聚碳酸酯。
可以对该反应中获得的氯化氢施以一个或多个提纯步骤以使其适用于许多其它的用途可能性,特别是用于电化学氧化或热氧化以形成氯气。由此获得的这种氯气可以用于与一氧化碳制备光气;所得光气可用于本发明的方法。
在反应条件下为液体的最终产物在包含步骤b)、c)和e)的多个分离步骤中与副产物和催化剂或其HCl加合物分离。其随后优选具有大于95%,优选大于99.0%,特别优选大于99.5%的碳酸二芳基酯和任选的酚的含量。该最终产物优选主要包含碳酸二芳基酯。将该反应中所用的催化剂后处理,以使其可至少部分再循环至该反应(步骤d)。
本发明的方法由三个方法区段构成:
I. 包含方法步骤a)的反应,
II. 氯化氢后处理(任选),
III. 通过蒸馏提纯产物和分离催化剂,其包含方法步骤b)、c)和e),其中回收的催化剂至少部分再循环至阶段I(步骤d))。
在方法区段I),即反应中,在上游的方法步骤中将反应物互相混合,以存在光气在熔融单酚中的基本均匀溶液;这可任选在预定的熔体温度下通过使用升高的压力实现。
本发明中制备的碳酸二芳基酯优选是通式(I)的那些
(I)
其中R、R'和R''可以各自彼此独立地为氢、卤素或支化或非支化的C1-至C9-烷基或支化或非支化的C1-至C9-烷氧基羰基。R、R'和R''优选在式(I)的两侧上相同。
特别优选的是碳酸二苯酯。
适用于本发明的单酚优选是通式(II)的那些
(II)
其中R、R'和R''可以各自彼此独立地具有对通式(I)所述的含义。
对本发明而言,“C1-C4-烷基”是例如甲基、乙基、正丙基、异丙基、正丁基、仲丁基、叔丁基;“C1-C6-烷基”另外是例如正戊基、1-甲基丁基、2-甲基丁基、3-甲基丁基、新戊基、1-乙基丙基、环己基、环戊基、正己基、1,1-二甲基丙基、1,2-二甲基丙基、1,2-二甲基丙基、1-甲基戊基、2-甲基戊基、3-甲基戊基、4-甲基戊基、1,1-二甲基丁基、1,2-二甲基丁基、1,3-二甲基丁基、2,2-二甲基丁基、2,3-二甲基丁基、3,3-二甲基丁基、1-乙基丁基、2-乙基丁基、1,1,2-三甲基丙基、1,2,2-三甲基丙基、1-乙基-1-甲基丙基、1-乙基-2-甲基丙基或1-乙基-2-甲基丙基;“C1-C9-烷基”另外是例如正庚基和正辛基或正壬基。这同样适用于烷基羰基中的相应烷基。
合适的单酚是例如:苯酚、烷基苯酚如甲酚、对-叔丁基苯酚、对-枯基苯酚、对-正辛基苯酚、对-异辛基苯酚、对-正壬基苯酚和对-异壬基苯酚、卤代苯酚如对-氯苯酚、2,4-二氯苯酚、对-溴苯酚、2,4,6-三溴苯酚,苯甲醚和水杨酸甲酯或水杨酸苯酯。
特别优选的是苯酚。
所用单酚应具有至少99.90重量%的纯度。
反应物优选含有少于300体积ppm的水,因为水的存在促进该装置材料的腐蚀。
此处所用的单酚可以除了含有从外部引入整个工艺的酚,即来自储罐的所谓的新鲜酚,还含有来自方法步骤II)和III)的冷凝物料流或来自方法步骤II)的洗涤液体料流的再循环的单酚。这样再循环的单酚可含有不破坏该反应的来自该方法的副产物,例如残留量的碳酸二芳基酯、氯化氢或氯碳酸芳基酯。单酚在所用的反应物混合物中优选以大于基于光气计的化学计算所需量存在。单酚与光气的摩尔比可以在1.5:1至4:1变化,优选的是2:1至3:1的摩尔比,特别优选的是2.5:1至3:1的摩尔比。
在下文中,术语“氯甲酸芳基酯”用于表示在由单酚和光气制备碳酸二芳基酯时作为中间产物形成的化合物。
适用于本发明的氯甲酸芳基酯优选是通式(III)的那些
(III)
其中R、R'和R''可以各自彼此独立地具有对通式(I)所述的含义。
如果通式(III)的氯甲酸芳基酯与通式(II)的单酚反应,R、R'和R''优选各自在式(I)和(II)中具有相同含义。
特别优选的是氯甲酸苯酯。
为避免该制备方法的最终产物中的不合意副产物,所用光气应具有至少99.80重量%,优选99.96重量%的纯度;四氯化碳含量应小于50体积ppm,优选小于15体积ppm。
根据本发明,使用取代或未取代的吡啶作为催化剂。其可以以游离碱的形式或完全或部分以其盐酸盐的形式存在。“以游离碱的形式”或“以游离形式”对本发明而言是指该吡啶环的氮不以质子化形式存在。
优选最多10摩尔%,特别优选最多1摩尔%的任选取代的吡啶以游离形式存在。其余部分以盐酸盐形式存在。
根据本发明充当催化剂的吡啶优选是通式(IV)的那些
(IV)
其中R1和R2可以各自彼此独立地为H、支化或非支化的C1-至C9-烷基、C5-或C6-环烷基、OH、OR3、NHR3或NR3R4,其中R3和R4各自彼此独立地为C1-至C4-烷基。特别优选的是R1和R2为H。
合适的吡啶是例如吡啶、2-甲基吡啶、3-甲基吡啶、4-甲基吡啶、2-乙基吡啶、3-乙基吡啶、4-乙基吡啶、2-异丙基吡啶、3-异丙基吡啶、4-异丙基吡啶、2-丁基吡啶、4-叔丁基吡啶、2,3-二甲基吡啶、2,4-二甲基吡啶、2,5-二甲基吡啶、2,6-二甲基吡啶、3,4-二甲基吡啶、3,5-二甲基吡啶、3,4-二乙基吡啶、3,5-二乙基吡啶、3-乙基-4-甲基吡啶、2-(3-戊基)吡啶、4-(3-戊基)吡啶、2-二甲基氨基吡啶、4-二甲基氨基吡啶、2-甲氧基吡啶、2,6-二甲氧基吡啶、4-环己基吡啶、4-(5-壬基)吡啶、4-苯基丙基吡啶和2-羟基吡啶。
特别优选的是吡啶。
在一个特别优选的实施方案中,该催化剂是吡啶盐酸盐。
根据本发明使用的催化剂可以以基于存在的单酚计0.001摩尔%至10摩尔%的量,优选0.01摩尔%至5摩尔%的量使用。
该催化剂以在单酚熔体中的溶液的形式使用。这种溶液根据本发明含有从方法区段III)经过或未经过单独的催化剂后处理而再循环到作为方法区段I)的反应中的催化剂的至少部分量。对于将催化剂再循环到方法区段I)中,催化剂后处理因此不是绝对必要的,而是完全可能的。
最早在反应物完全充分混合后,优选在反应器中进行催化剂的添加,以避免反应物在混合过程中过早反应和因此在不合适的方法区段中过早生成氯化氢。
来自方法区段III)的催化剂的再循环可以任意频繁地进行;在连续方法中,可优选连续再循环部分量的催化剂,而任选从工艺循环中排出部分量,以防止催化剂污染或任选虑及催化剂的失活。在需要时,可以将新鲜催化剂添加到再循环量的催化剂中。在一个优选实施方案中,再循环至少25重量%,特别优选至少50重量%,非常特别优选至少75重量%,特别是非常特别优选至少85重量%的催化剂。但是,在一个优选实施方案中,再循环最多99重量%,优选最多95重量%的催化剂。
反应物单酚和光气以上示摩尔比或以上述优选摩尔比互相混合,其中单酚始终作为熔体存在,且光气根据主导压力为气体或液体。在常压和高于60℃的温度下,主要存在双相的气液混合物,因为光气在单酚中的溶解度也如同在碳酸二芳基酯中一样随温度提高而降低。
因此,熔融单酚和光气的混合物必须在反应相中非常剧烈混合并再分散,以借助相界面的令人满意的补充确保反应物的充分反应。或者,在凝聚均相中可以显著提高光气与酚的反应(由于与气态光气和液态酚的两相混合物相比,光气在酚中的浓度提高)。提高温度也对反应速率具有加速作用,因此100℃至250℃,优选110℃至220℃的升高的温度可能是有利的。但是,由于如上文提到的这样的温度不利于光气在酚中的溶解度,在加压下在升高的温度下进行反应是特别有利的。因此,将反应物在升高的温度在常压下,优选在最多50巴(绝对)的升高的压力下,特别优选在最多30巴(绝对)的升高的压力下,非常特别优选在4至25巴(绝对)的压力下互相混合并反应。混合区中的温度应至少为单酚的熔点,但100℃至250℃的反应温度是有利的。
在反应物基本完成混合后,优选将上述催化剂之一优选以所述优选量作为在单酚中的溶液添加到该混合物中。由于单酚与光气形成氯碳酸芳基酯作为中间体的催化反应在上述温度和压力下极快进行并消去气态氯化氢,该反应优选通过多级方式进行。该反应可以在绝热条件下进行,因为其只有轻微的放热(Wärmetönung)。在第一阶段,即所谓的主反应器中,除已进一步反应的碳酸二芳基酯外主要形成氯碳酸芳基酯,特别是在升高的压力下和优选在120℃至230℃的温度下,特别优选在130℃至210℃的温度下,对于碳酸二苯酯的制备,非常特别优选在170℃至200℃的温度下,并且在15至120分钟,优选45至90分钟的反应器液体停留时间下。在第二阶段中,在所谓的后反应器中在优选170℃至250℃,特别优选190℃至230℃,非常特别优选200℃至210℃的略更高的温度下在15至120分钟,优选45至90分钟的反应器停留时间下,氯碳酸芳基酯与仍存在的单酚反应以形成碳酸二芳基酯。在此,也可以将第二阶段中在所谓的后反应器中的压力降低到2至20巴。压力的这种降低可以有利地在所谓的闪蒸阶段(Flash-Stufe)中进行,其中由于压力降低而可以特别好地从反应熔体中分离出在主反应器中形成的氯化氢气体。在后反应器中的第二反应阶段下游也可任选存在用于分离出残留量的氯化氢的闪蒸阶段。其也可任选集成到后续方法区段III)(通过蒸馏的产物提纯和催化剂分离)的第一蒸馏塔中,并在此加速气相和液相的分离。
连续反应器优选很好地适合作为用于反应物在所示反应条件下的反应的反应器,但也可以使用搅拌釜作为分批式反应器。特别好合适的连续反应器是例如搅拌釜级联、泡罩塔、板式塔、填料塔或具有用于充分混合反应介质的固定内装件的塔或反应蒸馏塔。
这样的塔也可互相组合,例如泡罩塔与上叠的精馏塔,在这种情况下不同于反应物的上述混合,可以在塔组合的不同位置处分开引入反应物。因此,例如,在上述塔组合的情况下,可以将光气引入下方的泡罩塔并将单酚与催化剂一起引入具有大约10个理论塔板的上方的精馏塔。从泡罩塔中取出形成的碳酸二芳基酯。
反应物的相应的分开计量加入也可以在反应蒸馏塔中如下进行,即通过在塔中部引入光气并在塔顶部与催化剂一起引入单酚。从塔底取出反应混合物。这样的塔可具有至少5个,优选大约20个塔板。
在反应器的另一任选实施方案中,反应物可以在主反应器中在1至25巴(绝对)的压力下在足够高的任选更长的停留时间下,但在反应器下部的优选120℃至190℃,特别优选160℃至180℃的较低温度下完全反应。在反应器上部必须另外加热,以在此实现最多250℃,优选最多230℃的略更高的温度。随后可以借助闪蒸或另一脱气技术进行反应混合物的基本脱气和低沸物的分离。
特别优选的是泡罩塔,如上所述的反应物混合物自下向上流经其中。在此,在泡罩塔的塔顶部取出气态氯化氢并在该塔轴(Kolonnen-Shaft)的上端取出反应混合物。将其经该塔的底部供入充当后反应器的下一泡罩塔。在停留反应器(Verweil-Reaktor)末端从最后一个泡罩塔中取出完全反应的反应混合物,并供入后续的方法区段III)(通过蒸馏的产物提纯和催化剂分离)。在每种情况下在泡罩塔的顶部取出的氯化氢气体在后续的方法区段II)(氯化氢后处理)中提纯。在各个阶段之间也可通过在闪蒸中的减压和随后的增压而另外分离氯化氢。
装置材料必须满足在高温下对氯化氢和光气的耐受性的高要求,并优选选自材料:黑钢、不锈钢、钢合金、镍基合金(例如Hastelloy C)、陶瓷、石墨、搪瓷涂覆的材料、PTFE包覆的材料。
任选的方法区段II)(氯化氢后处理)的目标是分离和提纯副产物氯化氢。为此,收集反应A)中形成的气相并将氯化氢气体与可任选再循环以进一步反应形成碳酸二芳基酯的其它组分分离。可以蒸馏副产物氯化氢以提高纯度。此外,可以混入来自方法区段III)的气态子流。
在这一方法区段II)中,合并来自方法区段I)的含HCl的料流并共同提纯。优选在此不中和氯化氢。在低沸点组分中的主要产物是94重量%或更多的氯化氢气体;副产物是大于3重量%的过量使用的单酚以及痕量的氯碳酸芳基酯、碳酸二芳基酯和光气和作为来自光气的副产物的痕量的一氧化碳和四氯化碳。可以借助各种步骤将副产物与主要产物氯化氢基本分离,以获得具有大于99.0体积%,优选大于99.8体积%的纯度的氯化氢气体和小于1000体积ppm,优选小于500体积ppm的残留含量的光气和/或氯碳酸酯。氯化氢中的有机化合物含量同样应小于1000体积ppm,优选小于50体积ppm;特别地,含氯烃的含量应小于50体积ppm。
通过一个或多个下述步骤实现这一目的。优选通过多级法实现这一目的。优选通过蒸馏分离出氯化氢。
在所谓的第一冷凝阶段中,在合适的温度下部分冷凝出具有比氯化氢更高的沸点的副产物。在此,特别是从氯化氢气体中基本除去以较高浓度存在的较高沸点组分,例如单酚和碳酸二芳基酯,并再循环至该反应。当除了较低温外还任选使用升高的压力时,这种分离特别成功。第一冷凝阶段中的优选温度为至少80℃,并且对于碳酸二苯酯的制备而言特别优选90℃。优选将压力设定为8至25巴(绝对),用于制备碳酸二苯酯的特别优选的压力为12巴(绝对)。也可任选通过多级方式在各种温度和/或压力下从氯化氢气体流中冷凝副产物。
如果足够低的温度或足够高的压力在技术上不可实现或难以实现,也可以跳过这种第一冷凝阶段,以在后续的所谓的HCl洗涤阶段中在合适的装置中使用熔融碳酸二芳基酯从氯化氢料流中洗除出副产物。如果这一HCl洗涤阶段是避开第一冷凝阶段的氯化氢的第一提纯阶段,这一HCl洗涤阶段也可本身通过多级进行并在各种降低的温度水平下运行,以提高该洗涤的效率。在此,单酚特别地极易溶于碳酸二芳基酯。当例如在后续的方法区段C)(碳酸二芳基酯后处理)中的合适位置取出用于该洗涤的碳酸二芳基酯时,痕量的氯碳酸酯和光气也可在这一方法步骤中反应形成碳酸二芳基酯。原则上,从这一方法区段到经蒸馏的碳酸二芳基酯的各个碳酸二芳基酯料流都适用于该HCl洗涤阶段,并且对上述有机氯化合物的反应有利的是从方法区段III)中取出用于HCl洗涤阶段的含催化剂和酚的碳酸二芳基酯料流,以能使仍存在于氯化氢气体中的有机氯化合物在短时间内反应。
这样的合适碳酸二芳基酯是离开方法区段I)(反应)并为进一步后处理而供入方法区段III)的第一阶段(碳酸二芳基酯后处理)的粗制碳酸二芳基酯。在这种碳酸二芳基酯中存在足够量的催化剂和单酚。或者,经蒸馏的碳酸二芳基酯可以以任意方式用于HCl洗涤阶段,因为要洗出的副产物在DPC中的物理溶解度足够高。但是,经蒸馏的纯碳酸二芳基酯优选用于HCl洗涤阶段。为了在HCl洗涤阶段中使有机氯化合物反应,同样也可以使用单酚代替碳酸二芳基酯作为洗涤介质,因为要洗出的副产物在单酚中的物理溶解度也足够高。这种单酚可以是例如单酚反应物料流的子流。如果需要含氯酯(Chlorester)或光气反应以形成碳酸二芳基酯,用于该洗涤的单酚可以以任意方式含有催化剂。使用碳酸二芳基酯或使用单酚的HCl洗涤优选在高于碳酸二芳基酯的熔点的温度下进行;在碳酸二苯酯的制备中,80-95℃的熔融温度特别优选。该HCl洗涤可以在常压下或8至25巴(绝对)的升高的压力下进行;在碳酸二苯酯的制备中,12巴(绝对)特别优选。
在这种洗涤中,可以获得具有大于99.8重量%的纯度的氯化氢气体。优选地,光气的含量低于500体积ppm,氯甲酸酯的含量低于检出限,并将酚的含量降至低于10体积ppm。
这一HCl洗涤阶段不是绝对必要的,并在其它方法步骤的任意互相组合的情况下也可避开。
氯化氢蒸馏特别好地适用于实现氯化氢气体的高纯度。为了能以能量有效的方式进行这样的蒸馏,在上游的第二冷凝阶段中将待提纯的氯化氢预先冷却至较低温是有用但不是绝对必要的。如果省略这一阶段,在后续的氯化氢蒸馏中需要在低温下的相应较高能量。在也可任选在多种不同温度和/或压力水平下运行的这种第二冷凝阶段中,分离出仍包含于氯化氢气体中的痕量的较高沸点副产物,特别是在使用8至25巴(绝对)的较高压力时,在碳酸二苯酯的情况下优选12巴(绝对)时。该温度可根据技术情况在加25℃至减50℃的极宽范围内变化。当已使用单酚进行HCl洗涤阶段中的洗涤时,这种第二冷凝阶段特别值得推荐,因为由此可显著降低该HCl气体流中存在的单酚浓度并由此降低HCl蒸馏中的负担。如果省略这种第二冷凝阶段,对HCl蒸馏中的能量需求的要求相应地远远更高。冷凝物同样可以如第一冷凝阶段中那样供入该反应。
作为方法区段II)中的氯化氢后处理的第四和最后阶段,氯化氢的蒸馏在一个特别优选的实施方案中特别好地适用于制备高纯度的氯化氢。其优选应在升高的压力下进行,因为否则用于设定替代性所需的足够低温的能量消耗会不合比例地高。如果之前的提纯阶段已在常压下进行,最晚在这一提纯阶段中将氯化氢料流压缩到8至25巴(绝对)的较高压力是非常值得推荐的;对于碳酸二苯酯的制备,12巴(绝对)特别优选。在这些条件下可获得具有99.95重量%的纯度的氯化氢气体。
方法区段II)中的氯化氢提纯的所有四个上述阶段在所述顺序下特别好地适合根据本发明用于制备高纯度的氯化氢气体。不是绝对必须遵守特定的顺序或实施所有方法阶段,而是取决于从反应中分离出的氯化氢的污染程度和作为最终产物的氯化氢气体的所需纯度。因此,如下文以HCl蒸馏为例所示,使用各个提纯阶段或单个提纯阶段就任选完全可能实现所需结果。
如果来自方法区段I)(反应)的进料流没有预先提纯就直接供入氯化氢蒸馏,在相同温度和压力条件下同样可获得具有99.95重量%的纯度的氯化氢气体。
提纯阶段的组合完全有可能以不依赖于上述列举内容的特定顺序进行以实现特定纯度。
作为用于进行第一和第二冷凝阶段的装置,具有对工艺条件而言足够高的热交换器表面积和用于将冷凝物进给到该反应中的装置的经典冷阱是合适的。这样的冷阱也可制造成多级的并任选不同调温的。适用于HCl洗涤阶段的装置特别是连续运行的装置,如泡罩塔、泡罩板式塔、填料塔、填充塔(Packungskolonnen)、具有固定内装件的塔,其中洗涤液体可以从上方与上行氯化氢气体逆向传送。连续运行的搅拌装置,例如混合器-沉降器,或不连续运行的搅拌装置原则上也合适。
氯化氢蒸馏可以在具有合适的塔内装件的常规蒸馏塔或精馏塔中进行。
用于上述装置的材料必须满足对氯化氢的耐受性的高要求并优选选自黑钢、不锈钢、钢合金、镍基合金(例如Hastelloy C)、陶瓷、石墨、搪瓷涂覆的材料、PTFE包覆的材料。
在方法区段III)(产物提纯和催化剂分离)中,收集在反应I)中形成的较高沸点组分,分离并将催化剂以游离碱形式或以盐酸盐形式再循环至该反应。由此将主要产物提纯至获得具有大于99.0重量%,优选大于99.8重量%、特别优选大于99.95重量%的纯度的碳酸二芳基酯的程度。
已经令人惊讶地发现,可以借助蒸馏从反应混合物中分离和再循环催化剂,而不发生该催化剂的凝华。
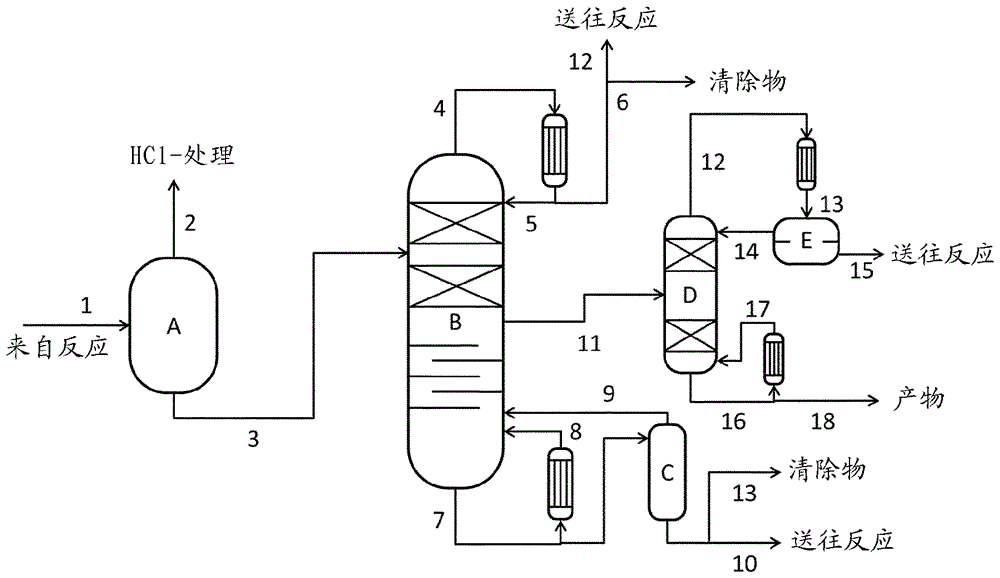
这一方法区段的图示综述显示在图1中。
在液体反应混合物的第一分离步骤中,在脱气阶段中基本分离出溶解的氯化氢。这可以借助闪蒸器(图1中的A)、蒸馏塔、这些装置的组合或另一常规的脱气技术(例如汽提)实现。
优选使用闪蒸阶段(A),其中通过降低压力将溶解的氯化氢脱气。在此选择20毫巴至1巴(绝对)的压力和140℃-205℃的温度,优选0.1巴至1巴(绝对)的压力和165-205℃的温度,特别优选0.3-0.7巴(绝对)的压力和180-200℃的温度。
或者,可以将在200毫巴至2巴(绝对),优选0.5至1巴(绝对),特别优选0.8-1巴(绝对)的压力下运行的蒸馏塔用于氯化氢的分离。
在闪蒸器的气相中或在替代性的蒸馏塔的顶部获得氯化氢、单酚和游离的任选取代的吡啶的混合物。优选将这一混合物在方法区段B)中添加到用于气体处理的主要气体料流中。
闪蒸器或替代性的塔的底部产物基本不含氯化氢,并在优选变体中单酚含量低。该底部产物因此由碳酸二芳基酯、单酚、游离形式或盐酸盐形式的任选取代的吡啶和副产物形成。
在另外的变体中,闪蒸阶段和蒸馏塔可以组合用于分离氯化氢或可以使用另一脱气技术(例如汽提)。或者,也可以省略第一分离步骤。但是这不优选,因为由此对于第二分离步骤产生较大的工艺料流,并且运送到进一步分离阶段中的氯化氢可能造成腐蚀问题。
在第二分离步骤中,通过分离出单酚、吡啶盐酸盐和次要组分从第一分离步骤的预提纯的料流(图1中的料流3)中分离出碳酸二芳基酯。在实验中已经发现,由碳酸二芳基酯和吡啶盐酸盐构成的二元混合物的蒸气-液体平衡具有非均相共沸行为(见图2)。在85重量%的吡啶-HCl的质量含量下测得最低共沸物。该液体分离成含吡啶盐酸盐的相和含碳酸二芳基酯的相。因此看起来难以借助其原理基于蒸气-液体平衡的分离技术,例如蒸馏将吡啶盐酸盐与碳酸二芳基酯分离。此外,140-146℃的吡啶盐酸盐的高熔点在蒸馏中可能构成阻碍,因为在冷凝器中的再升华会造成操作问题。
令人惊讶地,在实验中已证实通过蒸馏将碳酸二芳基酯与酚、吡啶盐酸盐和次要组分分离的可行性。
在该优选变体中,在第二分离步骤中提纯的碳酸二芳基酯因此在蒸馏塔的侧流(11)处获得(见图1)。在馏出物中取出单酚、游离吡啶、吡啶盐酸盐和低沸点次要组分。由于碳酸二芳基酯和吡啶盐酸盐之间的共沸物,该馏出物还具有大约5-15重量%的碳酸二芳基酯含量。
在塔底取出高沸点次要组分和热分解产物。为了在塔底不超过220℃的破坏产物的温度并使塔底产物保持可泵送,优选在塔底产物中保留10-50重量%,特别优选20-40重量%的碳酸二芳基酯含量。
在蒸馏塔的顶部优选设定5-100毫巴(绝对),特别优选10-40毫巴(绝对)的压力。根据顶部压力、单酚的过量和该反应中的催化剂浓度,顶部温度在60℃和140℃之间变化。在底部,以180-250℃,优选200-220℃的温度为主导。由于底部的这些高温,通过热分解降低次要组分水杨酸苯酯(Salol)的含量。由此可以在侧流中获得具有大于99重量%的纯度的碳酸二芳基酯。优选从蒸气相中取出该侧流,但也可从液相中取出。
可以使用侧塔进一步提高侧流中的碳酸二芳基酯纯度。但是,优选运行该蒸馏塔以使侧流不经进一步提纯就可引入后续的方法步骤,例如引入熔体聚碳酸酯方法中。
将该馏出物再循环至该反应。优选将馏出物的一部分作为清除物输出以排出低沸点次要组分。弃置该塔底产物或在一个优选变体中在除去高沸点清除物料流后再循环至该反应。在一个特别优选的变体中,在进一步分离操作(图1中的C),例如薄膜蒸发器中温和地分离出该塔底产物中包含的碳酸二芳基酯的一部分并重新送回该蒸馏塔,以降低高沸点清除物中的碳酸二芳基酯含量。
上述优选蒸馏的替代性变体安排在蒸馏塔的侧流中取出由碳酸二芳基酯和吡啶盐酸盐构成的混合物(见图3)。在附加蒸馏塔中,随后在底部从这种混合物中获得纯碳酸二芳基酯。
在该替代性变体的第一蒸馏塔(图3中的B)的顶部,在5-100毫巴(绝对),优选10-40毫巴(绝对)的顶部压力和60-115℃的顶部温度下取出酚和低沸点次要组分。塔底产物的组成和温度类似于蒸馏塔的上述优选变体中的塔底产物的。如上述优选变体中那样,将馏出物和塔底产物再循环至该反应。
在该替代性变体中用于提纯侧流的附加蒸馏塔(图3中的D)优选制造为非均相共沸蒸馏。顶部的滗析器(图3中的E)将该馏出物分成富含碳酸二芳基酯的相和富含吡啶盐酸盐的相。作为塔顶产物取出富含吡啶盐酸盐的相并再循环至该反应。将富含碳酸二芳基酯的相作为回流送回该塔。为了增加该回流,也可以将来自滗析器的两个相的混合物再循环至该塔。在塔底获得具有大于99重量%的纯度的碳酸二芳基酯,其不经进一步提纯就可送往后续的方法步骤,例如熔体聚碳酸酯法中。
附图解释:
图1显示方法区段III)(通过蒸馏的产物提纯和催化剂分离)的一个优选实施方案。
图2显示在各种温度(160℃、170℃和180℃)下碳酸二苯酯/吡啶盐酸盐混合物的实验测定的蒸气-液体-液体平衡 vs 压力。这在85重量%吡啶盐酸盐的质量含量下表现出最低共沸物。
图3描述方法区段III)(通过蒸馏的产物提纯和催化剂分离)的另一优选实施方案。
图1和3的解释:
A: 闪蒸阶段
B: 蒸馏塔
C: 附加分离操作(例如薄膜蒸发器)
D: 侧流蒸馏塔
E: 滗析器
1: 反应混合物
2: 来自闪蒸阶段的蒸气
3: 来自闪蒸阶段的底部产物
4: 来自蒸馏塔的蒸气
5: 返回蒸馏的回流
6: 再循环至该反应的馏出物
7: 来自蒸馏塔的塔底产物
8: 进入蒸馏塔的蒸发器蒸气
9: 来自附加分离操作(例如薄膜蒸发器)进入蒸馏塔的蒸气
10: 再循环至该反应的塔底产物
11: 产物料流(DPC)
12: 用于除去低沸点次要组分的清除物
13: 用于除去高沸点次要组分的清除物。
下列实施例旨在例示用于通过蒸馏分离吡啶盐酸盐的可行性的程序,但不构成限制。
实施例:
将碳酸二苯酯(DPC)、苯酚、吡啶盐酸盐(氯化吡啶鎓,下文也称作“吡啶·HCl”或“Py.HCl”)和Salol(水杨酸苯酯,苯酚的直接光气化的副产物)的各种混合物引入由具有30厘米长维格罗柱(包裹在铝箔中以隔热)的蒸馏烧瓶(1L)、李比希冷凝器(使用温水在80℃下运行)、冷阱(在-80℃下)和真空泵构成的实验室蒸馏装置中。测量柱顶部和底部的温度。熔融混合物在每种情况下在大约20毫巴(绝对)下分馏。单独收集2或3个馏分,并如同冷阱的底部产物和内容物一样通过气相色谱法分析。
实施例1:
在所述装置中蒸馏下列合成反应混合物:
在蒸馏中形成下列馏分:
令人惊讶地,馏分2在室温下是液体并也持久保持这样。
各馏分的分析得出下列组成(组成总量与100%的偏差归因于测量误差):
显而易见的是,各馏分中吡啶与HCl之间的摩尔比在每种情况下为大约1:1。相反,蒸馏柱底产物不含吡啶盐酸盐。在冷阱中,重新获得少量HCl,但没有以盐形式的吡啶盐酸盐沉积的迹象。这表明吡啶盐酸盐可令人惊讶地以盐形式作为低沸物从粗制碳酸二苯酯(DPC)反应混合物中蒸馏出。
实施例2:
在所述装置中蒸馏下列合成反应混合物:
在蒸馏柱中形成下列馏分:
由于显著低的苯酚含量,不同于实施例1,没有出现主要由苯酚构成的第一馏分。其它观察结果,特别是在室温下为液体的馏分2在实施例2的情况中也适用。
各馏分的分析得出下列组成(组成总量与100%的偏差归因于测量误差):
又显而易见的是,特别在馏分2(其中重新获得所用吡啶盐酸盐的主要部分)中,吡啶与HCl之间的摩尔比几乎理想地为1:1。该蒸馏柱底产物又不含吡啶盐酸盐。在冷阱中又重新获得少量HCl,但在这一实验中,也没有以盐形式的吡啶盐酸盐沉积的迹象。由此证实,吡啶盐酸盐可令人惊讶地以盐形式作为低沸物从粗制碳酸二苯酯(DPC)反应混合物中蒸馏出。
对比例1:
作为对比例,蒸馏吡啶盐酸盐的下列混合物(含有痕量水):
在蒸馏中形成下列馏分:
各馏分的分析得出下列组成(组成总量与100%的偏差归因于测量误差):
在加热纯吡啶盐酸盐时,在所选温度下也几乎没有发生该盐的离解。仅在馏分1中,温度、在室温下的物质状态以及组成都表明消去纯吡啶。
在馏分2中,发现少量的大约1:1组成的吡啶·HCl。
吡啶盐酸盐的主要部分如预期那样留在柱底产物中。在冷阱中又重新获得少量HCl,但在这一实验中也没有以盐形式的吡啶盐酸盐沉积的迹象。
对比例2:
制造40重量%吡啶盐酸盐和60重量%苯酚的合成混合物(类似于来自实施例1和2的馏分2)。该混合物在室温下为液体。溶解在苯酚中的吡啶盐酸盐的凝固点的降低导致在该蒸馏中出现的混合物也为液体,并因此可预计在工业蒸馏装置中也没有在真空管路中等的凝华。
- 还没有人留言评论。精彩留言会获得点赞!