神经活性类固醇、组合物、及其用途的制作方法
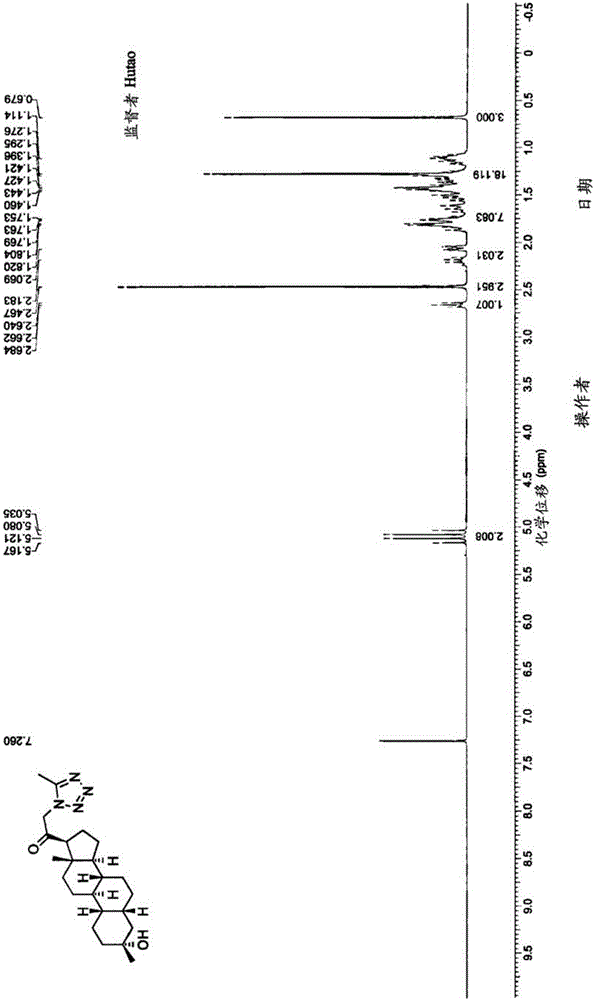
本申请要求于2014年5月29日提交的国际申请号PCT/CN2014/078820的优先权,其全部内容通过引用并入本文。发明背景脑兴奋性(excitability)被定义为动物的唤醒水平,是范围从昏迷到惊厥的连续体,并通过各种神经递质调节。通常,神经递质负责调节离子跨越神经元膜的传导性。在静止时,神经元膜具有约-70mV的电势(或膜电压),细胞内部相对于细胞外部是负的。电势(电压)是跨越神经元半透膜的离子(K+、Na+、Cl-、有机阴离子)平衡的结果。神经递质存储在突触前泡囊中,并且在神经元动作电位的影响下释放。当释放到突触间隙中时,兴奋性化学递质(例如,乙酰胆碱)将导致膜去极化(电势从-70mV到-50mV的变化)。该作用通过突触后的烟碱受体介导,所述突触后的烟碱受体由乙酰胆碱刺激以增加Na+离子的膜渗透性。降低的膜电势以突触后动作电位的形式刺激神经元兴奋性。在GABA受体复合物(GRC)的情况下,对脑兴奋性的作用通过GABA神经递质介导。GABA对总的脑兴奋性具有深度的影响,这是因为脑中的高达40%的神经元利用GABA作为神经递质。GABA通过调节氯离子跨越神经元膜的传导来调节单独的神经元的兴奋性。GABA与其在GRC上的识别位点相互作用,促进氯离子沿着GRC的电化学梯度流入到细胞中。该阴离子水平的细胞内增加导致跨膜电位的超极化,使神经元对于兴奋性输入不太敏感(即,神经元兴奋性的降低)。换句话说,神经元中的氯离子浓度越高,脑兴奋性(唤醒水平)越低。有许多文献证明,GRC负责介导焦虑、发作行为和镇静。因此,GABA和像GABA一样起作用或促进GABA的作用的药物(例如,治疗上使用的巴比妥酸盐和苯并二氮(BZ),例如),通过与GRC上的特异性调节位点相互作用,产生其治疗上有用的效果。积累的证据现已表明,除苯并二氮和巴比妥酸盐结合位点之外,GRC还包含对于神经活性类固醇的独特的位点(Lan,N.C.等人,Neurochem.Res.16:347-356(1991))。神经活性类固醇可内源性地出现。最有效的内源性神经活性类固醇为3α-羟基-5-还原孕甾烷-20-酮和3α-21-二羟基-5-还原孕甾烷-20-酮,其分别为激素类固醇孕酮和脱氧皮质酮的代谢物。这些类固醇代谢物改变脑兴奋性的能力在1986年被认识到(Majewska,M.D.等,Science232:1004-1007(1986);Harrison,N.L.等,JPharmacol.Exp.Ther.241:346-353(1987))。卵巢激素孕酮及其代谢物已被证明对脑兴奋性有深度的影响(Backstrom,T.等,ActaObstet.Gynecol.Scand.Suppl.130:19-24(1985);Pfaff,D.WandMcEwen,B.S.,Science219:808-814(1983);Gyermek等,JMedChem.11:117(1968);Lambert,J.等,TrendsPharmacol.Sci.8:224-227(1987))。孕酮及其代谢物的水平随着月经周期的阶段而改变。已有许多文献证明,孕酮及其代谢物的水平在月经开始之前降低。每月一次重现的在月经开始之前的一些身体症状也已被许多文献证明。已变成与经前综合征(PMS)有关的这些症状包括应激(紧张)、焦虑和偏头痛(Dalton,K.,PremenstrualSyndromeandProgesteroneTherapy,2ndedition,ChicagoYearbook,Chicago(1984))。具有PMS的受试者具有每月一次重现的在经期前存在且在经期后不存在的症状。以类似的方式,孕酮的减少也已与女性癫痫患者即月经性癫痫的发作频率的增加在时间上有关(Laidlaw,J.,Lancet,1235-1237(1956))。对于孕酮代谢物的减少,已观察到更直接的关联(Rosciszewska等,J.Neurol.Neurosurg.Psych..49:47-51(1986))。另外,对于具有原发性全身性小发作癫痫的受试者,发作的时间发生率已与经前综合征的症状的发生率有关(Backstrom,T.等,J.Psychosom.Obstet.Gynaecol..2:8-20(1983))。已发现类固醇脱氧皮质酮在治疗具有与其月经周期有关的癫痫发作的受试者方面是有效的(Aird,R.B.andGordan,G.,J.Amer.Med.Soc.145:715-719(1951))。还与低的孕酮水平有关的综合征是产后抑郁症(PND)。在分娩之后,孕酮水平立即急剧降低,导致PND的发作。PND的症状范围从轻度抑郁症到需要住院治疗的精神病。PND也与重度焦虑和应激性(irritability)有关。PND有关的抑郁症不能通过经典的抗抑郁药治疗,且经历PND的女性显示PMS的增加的发生率(Dalton,K.,PremenstrualSyndromeandProgesteroneTherapy,2ndedition,ChicagoYearbook,Chicago(1984))。共同地,这些观察结果暗示孕酮和脱氧皮质酮且更特别是它们的代谢物在脑兴奋性的稳态调节中的关键作用,其显示为与月经性癫痫、PMS和PND有关的发作行为或症状的增加。降低的孕酮水平和与PMS、PND和月经性癫痫有关的症状之间的相关性(Backstrom,T.等,JPsychosom.Obstet.Gynaecol.2:8-20(1983));Dalton,K.,PremenstrualSyndromeandProgesteroneTherapy,2ndedition,ChicagoYearbook,Chicago(1984))已促使将孕酮用在其治疗中(Mattson等,“Medroxyprogesteronetherapyofcatamenialepilepsy,”AdvancesinEpileptology:XVthEpilepsyInternationalSymposium,RavenPress,NewYork(1984),pp.279-282和Dalton,K.,PremenstrualSyndromeandProgesteroneTherapy,2ndedition,ChicagoYearbook,Chicago(1984))。然而,孕酮在上述综合征的治疗中不是一贯地有效的。例如,对于在PMS的治疗中的孕酮,剂量-响应关系不存在(Maddocks等,Obstet.Gynecol.154:573-581(1986);Dennerstein等,Brit.MedJ290:16-17(1986))。需要用作脑兴奋性的调节剂的新的和改善的神经活性类固醇,以及用于CNS相关疾病的预防和治疗的药物。本文中描述的化合物、组合物和方法针对该目标。发明概述本发明部分地基于提供具有良好的效力、药代动力学(PK)性能、口服生物利用度、可配制性(formulatability)、稳定性、安全性、清除率和/或代谢的新的19-去甲(即,C19去甲基)化合物的期望。本文描述的化合物的一个关键特征是在C3位的二取代(例如,一个取代基是3α羟基部分)。本发明人发现,在C-3处的二取代可消除羟基部分氧化为酮的可能性,防止进一步的代谢,并降低次级消除途径(例如,葡萄苷酸化)的可能性。本发明人进一步发现,C3二取代的总效果应该改善总的PK参数,并降低潜在的毒性和副作用,在一些实施方式中,这可以容许口服和/或长期施用。本文描述的化合物的另一关键特征是在C19位存在氢(“19-去甲”),而不是甲基。本发明人发现,19-去甲化合物与其C19-甲基对应物相比,具有改善的物理性质,例如,改善的溶解度。本发明人发现溶解度的进一步增加,例如,当AB环体系处于顺式构型时。因此,一方面,本文提供式(I)的19-去甲C3-3-二取代的C21-三唑和四唑类固醇及其药学可接受盐:其中A选自下组:R1是C1-C6卤代烷基(CHF2,CH2F)或C1-C6烷基(例如CH3,CH2CH3,杂烷基,例如CH2OCH3,CH2OCH2CH3);R2和R3独立选自:H,卤素(例如F),C1-C6烷基(例如CH3)或烷氧基(例如OCH3,OCH2CH3);R4是卤素(例如Cl,F),氰基,硝基,-S(O)xRa,-NRbRc,C1-C6烷基(例如CH3,CF3),C1-C6烷氧基,-C(O)Ra,-C(O)ORa,或-C(O)NRbRc;Ra是H或C1-C6烷基;每个Rb和Rc独立是H,-S(O)xRa,-C(O)Ra,C1-C6烷基,或C1-C6烷氧基,或Rb和Rc与它们附接的原子一起形成环;n是0至2的整数;并且x是0至2的整数;其中当A是(A-1)或(A-2)时,则R1选自:-CHF2,-CH2F,-CCl3,-CHCl2,-CH2Cl,-CBr3,-CHBr2,-CH2Br,或C1-C6烷基;或当A是(A-3)或(A-5)时,R1是–CH3,-CH2F,-CH2OCH3,或–CHF2,并且n是0,则R2和R3中的至少一个不是H。式(I)的类固醇,其亚属及其药学可接受盐在本文中统称为“本发明的化合物”。在另一个方面,提供了含有本发明的化合物和药用赋形剂的药物组合物。在一些实施方案中,本发明的化合物以有效量提供在所述药物组合物中。在一些实施方案中,本发明的化合物以治疗有效量提供。在一些实施方案中,本发明的化合物以预防有效量提供。在一些实施方案中,本文描述的化合物充当GABA调节剂,例如,以正的或负的方式影响GABAA受体。作为中枢神经系统(CNS)的兴奋性的调节剂,当通过其调节GABAA受体的能力介导时,预期这样的化合物具有CNS活性。因此,在另一个方面,提供了治疗需要其的受试者中的CNS相关病症的方法,所述方法包括:对受试者施用有效量的本发明的化合物。在一些实施方案中,所述CNS相关病症选自:睡眠障碍、心境障碍、精神分裂症谱系障碍、痉挛性障碍、记忆障碍和/或认知障碍、运动障碍、人格障碍、自闭症谱系障碍、疼痛、外伤性脑损伤、血管疾病、物质滥用障碍和/或戒断综合征,以及耳鸣。在一些实施方案中,口服、皮下、静脉内或肌肉内施用所述化合物。在一些实施方案中,长期施用所述化合物。由随后的详细说明、实施例和权利要求,其它目的和优点将对于本领域技术人员变得明晰。定义化学定义下面更详细地描述具体官能团和化学术语的定义。化学元素根据HandbookofChemistryandPhysics第75版的封面内页的CAS版本的元素周期表确定,且具体官能团通常如其中所描述的那样定义。另外,有机化学的一般原理,以及具体官能部分和反应性描述于如下中:ThomasSorrell,OrganicChemistry,UniversityScienceBooks,Sausalito,1999;SmithandMarch,March'sAdvancedOrganicChemistry,5thEdition,JohnWiley&Sons,Inc.,NewYork,2001;Larock,ComprehensiveOrganicTransformations,VCHPublishers,Inc.,NewYork,1989;和Carruthers,SomeModernMethodsofOrganicSynthesis,3rdEdition,CambridgeUniversityPress,Cambridge,1987。本文描述的化合物可包括一个或多个不对称中心,且因此可以存在多种异构体形式,例如,对映异构体和/或非对映异构体形式。例如,本文描述的化合物可为单独的对映异构体、非对映异构体或几何异构体,或者可为立体异构体的混合物的形式,包括外消旋混合物和富含一种或多种立体异构体的混合物。异构体可通过本领域技术人员已知的方法从混合物中分离,所述方法包括:手性高压液相色谱法(HPLC)以及手性盐的形成和结晶;或者优选的异构体可通过不对称合成来制备。参见,例如,Jacques等,Enantiomers,RacematesandResolutions(WileyInterscience,NewYork,1981);Wilen等,Tetrahedron33:2725(1977);Eliel,StereochemistryofCarbon化合物(McGraw-Hill,NY,1962);和Wilen,TablesofResolvingAgentsandOpticalResolutionsp.268(E.L.Eliel,Ed.,Univ.ofNotreDamePress,NotreDame,IN1972)。本发明另外包括作为基本上不含其它异构体的单独异构体、或者作为多种异构体的混合物的本文描述的化合物。当列出数值范围时,它包括每个值和在所述范围内的子范围。例如“C1-6烷基”包括C1、C2、C3、C4、C5、C6、C1-6、C1-5、C1-4、C1-3、C1-2、C2-6、C2-5、C2-4、C2-3、C3-6、C3-5、C3-4、C4-6、C4-5和C5-6烷基。下列术语意图具有下面随其提供的含义,并且在理解本发明的说明书和预期范围中是有用的。当描述本发明时,可以包括化合物、含有该化合物的药物组合物,以及该化合物和组合物的试验方法,下列术语,如果存在的话,具有下列含义,除非另有陈述。还应该理解,当本文描述时,任何下面所定义的部分可以被许多取代基取代,而且相应的定义在下面列出的它们的范围内,包括这种取代部分。除非另作说明,否则,术语“取代”如下面所定义。应该进一步理解,当本文使用时,术语“基团”和“原子团”可以互换使用。冠词“一个(种)(a,an)”可在本文中用来指该冠词的语法对象的一个(种)或超过一个(种)(即,至少一个(种))。作为例子,“类似物”意指一个(种)类似物或超过一个(种)类似物。“烷基”是指具有1至20个碳原子的直链或支链饱和烃基团(“C1-20烷基”)。在一些实施方案中,烷基具有1至12个碳原子(“C1-12烷基”)。在一些实施方案中,烷基具有1至10个碳原子(“C1-10烷基”)。在一些实施方案中,烷基具有1至9个碳原子(“C1-9烷基”)。在一些实施方案中,烷基具有1至8个碳原子(“C1-8烷基”)。在一些实施方案中,烷基具有1至7个碳原子(“C1-7烷基”)。在一些实施方案中,烷基具有1至6个碳原子(“C1-6烷基”,本文还指的是“低级烷基”)。在一些实施方案中,烷基具有1至5个碳原子(“C1-5烷基”)。在一些实施方案中,烷基具有1至4个碳原子(“C1-4烷基”)。在一些实施方案中,烷基具有1至3个碳原子(“C1-3烷基”)。在一些实施方案中,烷基具有1至2个碳原子(“C1-2烷基”)。在一些实施方案中,烷基具有1个碳原子(“C1烷基”)。在一些实施方案中,烷基具有2至6个碳原子(“C2-6烷基”)。C1-6烷基的例子包括:甲基(C1)、乙基(C2)、正丙基(C3)、异丙基(C3)、正丁基(C4)、叔丁基(C4)、仲丁基(C4)、异丁基(C4)、正戊基(C5)、3-戊基(C5)、戊基(C5)、新戊基(C5)、3-甲基-2-丁基(C5)、叔戊基(C5)和正己基(C6)。烷基的其它例子包括正庚基(C7)、正辛基(C8),等。除非另作说明,否则,烷基的每个独立地任选被取代,即,未取代的(“未取代的烷基”)或被一个或多个取代基取代(“取代的烷基”);例如,1至5个取代基、1至3个取代基或1个取代基。在一些实施方案中,所述烷基是未取代的C1-10烷基(例如,-CH3)。在一些实施方案中,烷基是取代的C1-10烷基。常规烷基缩写包括:Me(-CH3)、Et(-CH2CH3)、iPr(-CH(CH3)2)、nPr(-CH2CH2CH3)、n-Bu(-CH2CH2CH2CH3)或i-Bu(-CH2CH(CH3)2)。本文使用的“亚烷基”、“亚烯基”和“亚炔基”分别指的是烷基、烯基和炔基的二价基。当具体“亚烷基”、“亚烯基”和“亚炔基”提供了碳的范围或数目时,应当理解,所述范围或数目是指碳在直链二价碳链中的范围或数目。“亚烷基”、“亚烯基”和“亚炔基”可以被一个或多个本文所描述的取代基取代,或是未取代的。“亚烷基”是指除去烷基的两个氢而形成二价基的烷基,并且可以是取代或未取代的亚烷基。未取代的亚烷基包括但不局限于:亚甲基(-CH2-)、亚乙基(-CH2CH2-)、亚丙基(-CH2CH2CH2-)、亚丁基(-CH2CH2CH2CH2-)、亚戊基(-CH2CH2CH2CH2CH2-)、亚己基(-CH2CH2CH2CH2CH2CH2-),等。示例性的取代的亚烷基,例如,被一个或多个烷基(甲基)取代的亚烷基,包括但不局限于:取代的亚甲基(-CH(CH3)-、-C(CH3)2-)、取代的亚乙基(-CH(CH3)CH2-、-CH2CH(CH3)-、-C(CH3)2CH2-、-CH2C(CH3)2-)、取代的亚丙基(-CH(CH3)CH2CH2-、-CH2CH(CH3)CH2-、-CH2CH2CH(CH3)-、-C(CH3)2CH2CH2-、-CH2C(CH3)2CH2-、-CH2CH2C(CH3)2-),等。“烯基”是指具有2至20个碳原子、一个或多个碳-碳双键(例如,1、2、3或4个碳-碳双键)以及任选一个或多个碳-碳叁键(例如,1、2、3或4个碳-碳叁键)的直链或支链烃基团(“C2-20烯基”)。在一些实施方案中,烯基不含有任何叁键。在一些实施方案中,烯基具有2至10个碳原子(“C2-10烯基”)。在一些实施方案中,烯基具有2至9个碳原子(“C2-9烯基”)。在一些实施方案中,烯基具有2至8个碳原子(“C2-8烯基”)。在一些实施方案中,烯基具有2至7个碳原子(“C2-7烯基”)。在一些实施方案中,烯基具有2至6个碳原子(“C2-6烯基”)。在一些实施方案中,烯基具有2至5个碳原子(“C2-5烯基”)。在一些实施方案中,烯基具有2至4个碳原子(“C2-4烯基”)。在一些实施方案中,烯基具有2至3个碳原子(“C2-3烯基”)。在一些实施方案中,烯基具有2个碳原子(“C2烯基”)。一个或多个碳-碳双键可以在内部(例如,在2-丁烯基中)或端部(例如,在1-丁烯基中)。C2-4烯基的例子包括:乙烯基(C2)、1-丙烯基(C3)、2-丙烯基(C3)、1-丁烯基(C4)、2-丁烯基(C4)、丁二烯基(C4),等。C2-6烯基的例子包括:上述的C2-4烯基,以及戊烯基(C5)、戊二烯基(C5)、己烯基(C6),等。烯基的其它例子包括:庚烯基(C7)、辛烯基(C8),辛三烯基(C8),等。除非另作说明,否则,烯基的每个独立地任选被取代,即,未取代的(“未取代的烯基”)或被一个或多个取代基取代(“取代的烯基”);例如,1至5个取代基、1至3个取代基或1个取代基。在一些实施方案中,烯基是未取代的C2-10烯基。在一些实施方案中,烯基是取代的C2-10烯基。“亚烯基”是指除去烯基的两个氢而形成二价基的烯基,并且可以是取代或未取代的亚烯基。示例性的未取代的二价亚烯基包括但不局限于:亚乙烯基(-CH=CH-)和亚丙烯基(例如,-CH=CHCH2-、-CH2-CH=CH-)。示例性的取代的亚烯基,例如,被一个或多个烷基(甲基)取代的亚烯基,包括但不局限于:取代的亚乙基(-C(CH3)=CH-、-CH=C(CH3)-)、取代的亚丙烯基(-C(CH3)=CHCH2-、-CH=C(CH3)CH2-、-CH=CHCH(CH3)-、-CH=CHC(CH3)2-、-CH(CH3)-CH=CH-、-C(CH3)2-CH=CH-、-CH2-C(CH3)=CH-、-CH2-CH=C(CH3)-),等。“炔基”是指具有2至20个碳原子、一个或多个碳-碳叁键(例如,1、2、3或4个碳-碳叁键)以及任选一个或多个碳-碳双键(例如,1、2、3或4个碳-碳双键)的直链或支链烃基团(“C2-20炔基”)。在一些实施方案中,炔基不含有任何双键。在一些实施方案中,炔基具有2至10个碳原子(“C2-10炔基”)。在一些实施方案中,炔基具有2至9个碳原子(“C2-9炔基”)。在一些实施方案中,炔基具有2至8个碳原子(“C2-8炔基”)。在一些实施方案中,炔基具有2至7个碳原子(“C2-7炔基”)。在一些实施方案中,炔基具有2至6个碳原子(“C2-6炔基”)。在一些实施方案中,炔基具有2至5个碳原子(“C2-5炔基”)。在一些实施方案中,炔基具有2至4个碳原子(“C2-4炔基”)。在一些实施方案中,炔基具有2至3个碳原子(“C2-3炔基”)。在一些实施方案中,炔基具有2个碳原子(“C2炔基”)。一个或多个碳叁键可以在内部(例如,在2-丁炔基中)或端部(例如,在1-丁炔基中)。C2-4炔基的例子包括但不限于:乙炔基(C2)、1-丙炔基(C3)、2-丙炔基(C3)、1-丁炔基(C4)、2-丁炔基(C4),等。C2-6烯基的例子包括:上述C2-4炔基,以及戊炔基(C5)、己炔基(C6),等。炔基的其它例子包括庚炔基(C7)、辛炔基(C8),等。除非另作说明,否则,炔基的每个独立地任选被取代,即,未取代的(“未取代的炔基”)或被一个或多个取代基取代(“取代的炔基”);例如,1至5个取代基、1至3个取代基或1个取代基。在一些实施方案中,炔基是未取代的C2-10炔基。在一些实施方案中,炔基是取代的C2-10炔基。“亚炔基”是指除去炔基的两个氢而形成二价基的直链炔基,并且可以是取代或未取代的亚炔基。示例性的二价亚炔基包括但不限于:取代或未取代的亚乙炔基、取代或未取代的亚丙炔基,等。本文使用的术语“杂烷基”是指本文所定义的烷基,在母链内,它进一步含有一或多个(例如,1、2、3或4个)杂原子(例如,氧、硫、氮、硼、硅、磷),其中,一个或多个杂原子在所述母体碳链内的相邻碳原子之间,和/或,一个或多个杂原子在碳原子和母体分子之间,即,在连接点之间。在一些实施方案中,杂烷基是指具有1至10个碳原子和1、2、3或4个杂原子的饱和基团(“杂C1-10烷基”)。在一些实施方案中,杂烷基是具有1至9个碳原子和1、2、3或4个杂原子的饱和基团(“杂C1-9烷基”)。在一些实施方案中,杂烷基是具有1至8个碳原子和1、2、3或4个杂原子的饱和基团(“杂C1-8烷基”)。在一些实施方案中,杂烷基是具有1至7个碳原子和1、2、3或4个杂原子的饱和基团(“杂C1-7烷基”)。在一些实施方案中,杂烷基是具有1至6个碳原子和1、2或3个杂原子的饱和基团(“杂C1-6烷基”)。在一些实施方案中,杂烷基是具有1至5个碳原子和1或2个杂原子的饱和基团(“杂C1-5烷基”)。在一些实施方案中,杂烷基是具有1至4个碳原子和1或2个杂原子的饱和基团(“杂C1-4烷基”)。在一些实施方案中,杂烷基是具有1至3个碳原子和1个杂原子的饱和基团(“杂C1-3烷基”)。在一些实施方案中,杂烷基是具有1至2个碳原子和1个杂原子的饱和基团(“杂C1-2烷基”)。在一些实施方案中,杂烷基是具有1个碳原子和1个杂原子的饱和基团(“杂C1烷基”)。在一些实施方案中,杂烷基是具有2至6个碳原子和1或2个杂原子的饱和基团(“杂C2-6烷基”)。除非另作说明,否则,杂烷基的每个独立地是未取代的(“未取代的杂烷基”)或被一个或多个取代基取代(“取代的杂烷基”)。在一些实施方案中,杂烷基是未取代的杂C1-10烷基。在一些实施方案中,杂烷基是取代的杂C1-10烷基。本文使用的术语“杂烯基”是指本文所定义的烯基,它进一步含有一或多个(例如,1、2、3或4个)杂原子(例如,氧、硫、氮、硼、硅、磷),其中,一个或多个杂原子在所述母体碳链内的相邻碳原子之间,和/或,一个或多个杂原子在碳原子和母体分子之间,即,在连接点之间。在一些实施方案中,杂烯基是指具有2至10个碳原子、至少一个双键和1、2、3或4个杂原子的基团(“杂C2-10烯基”)。在一些实施方案中,杂烯基具有2至9个碳原子、至少一个双键和1、2、3或4个杂原子(“杂C2-9烯基”)。在一些实施方案中,杂烯基具有2至8个碳原子、至少一个双键和1、2、3或4个杂原子(“杂C2-8烯基”)。在一些实施方案中,杂烯基具有2至7个碳原子、至少一个双键和1、2、3或4个杂原子(“杂C2-7烯基”)。在一些实施方案中,杂烯基具有2至6个碳原子、至少一个双键和1、2或3个杂原子(“杂C2-6烯基”)。在一些实施方案中,杂烯基具有2至5个碳原子、至少一个双键和1或2个杂原子(“杂C2-5烯基”)。在一些实施方案中,杂烯基具有2至4个碳原子、至少一个双键和1或2个杂原子(“杂C2-4烯基”)。在一些实施方案中,杂烯基具有2至3个碳原子、至少一个双键和1个杂原子(“杂C2-3烯基”)。在一些实施方案中,杂烯基具有2至6个碳原子、至少一个双键和1或2个杂原子(“杂C2-6烯基”)。除非另作说明,否则,杂烯基的每个独立地是未取代的(“未取代的杂烯基”)或被一个或多个取代基取代(“取代的杂烯基”)。在一些实施方案中,杂烯基是未取代的杂C2-10烯基。在一些实施方案中,杂烯基是取代的杂C2-10烯基。本文使用的术语“杂炔基”是指本文所定义的炔基,它进一步含有一或多个(例如,1、2、3或4个)杂原子(例如,氧、硫、氮、硼、硅、磷),其中,一个或多个杂原子在所述母体碳链内的相邻碳原子之间,和/或,一个或多个杂原子在碳原子和母体分子之间,即,在连接点之间。在一些实施方案中,杂炔基是指具有2至10个碳原子、至少一个叁键和1、2、3或4个杂原子的基团(“杂C2-10炔基”)。在一些实施方案中,杂炔基具有2至9个碳原子、至少一个叁键和1、2、3或4个杂原子(“杂C2-9炔基”)。在一些实施方案中,杂炔基具有2至8个碳原子、至少一个叁键和1、2、3或4个杂原子(“杂C2-8炔基”)。在一些实施方案中,杂炔基具有2至7个碳原子、至少一个叁键和1、2、3或4个杂原子(“杂C2-7炔基”)。在一些实施方案中,杂炔基具有2至6个碳原子、至少一个叁键和1、2或3个杂原子(“杂C2-6炔基”)。在一些实施方案中,杂炔基具有2至5个碳原子、至少一个叁键和1或2个杂原子(“杂C2-5炔基”)。在一些实施方案中,杂炔基具有2至4个碳原子、至少一个叁键和1或2个杂原子(“杂C2-4炔基”)。在一些实施方案中,杂炔基具有2至3个碳原子、至少一个叁键和1个杂原子(“杂C2-3炔基”)。在一些实施方案中,杂炔基具有2至6个碳原子、至少一个叁键和1或2个杂原子(“杂C2-6炔基”)。除非另作说明,否则,杂炔基的每个独立地是未取代的(“未取代的杂炔基”)或被一个或多个取代基取代(“取代的杂炔基”)。在一些实施方案中,杂炔基是未取代的杂C2-10炔基。在一些实施方案中,杂炔基是取代的杂C2-10炔基。本文使用的“亚烷基”、“亚烯基”、“亚炔基”、“杂亚烷基”、“杂亚烯基”和“杂亚炔基”分别指的是烷基、烯基、炔基、杂烷基、杂烯基和杂炔基的二价基。当具体“亚烷基”、“亚烯基”、“亚炔基”、“杂亚烷基”、“杂亚烯基”或“杂亚炔基”提供了碳的范围或数目时,应当理解,所述范围或数目是指碳在直链二价碳链中的范围或数目。“亚烷基”、“亚烯基”、“亚炔基”、“杂亚烷基”、“杂亚烯基”和“杂亚炔基”可以被一个或多个本文所描述的取代基取代,或是未取代的。“芳基”是指具有提供在芳族环系中的6-14个环碳原子和零个杂原子的单环或多环的(例如,双环或三环)4n+2芳族环体系(例如,具有以环状排列共享的6、10或14个π电子)的基团(“C6-14芳基”)。在一些实施方案中,芳基具有六个环碳原子(“C6芳基”;例如,苯基)。在一些实施方案中,芳基具有十个环碳原子(“C10芳基”;例如,萘基,例如,1-萘基和2-萘基)。在一些实施方案中,芳基具有十四个环碳原子(“C14芳基”;例如,蒽基)。“芳基”还包括这样的环系统,在这种环系统中,上述芳基环与一个或多个碳环基或杂环基稠合,其中,原子团或连接点在所述芳基环上,在这种情况下,碳原子的数目继续表示所述芳基环系统中的碳原子数目。典型的芳基包括但不限于衍生自以下的基团:醋蒽烯、苊烯、醋菲烯(acephenanthrylene)、蒽、薁、苯、屈、晕苯、荧蒽、芴、并六苯、己芬、己搭烯、不对称引达省、对称引达省、茚满、茚、萘、并八苯、辛芬、辛搭烯、卵苯、戊-2,4-二烯、并五苯、戊搭烯、戊芬、苝、非那烯、菲、苉、七曜烯、芘、皮蒽、玉红省、苯并菲和联三萘。具体地说,芳基包括苯基、萘基、茚基和四氢萘基。除非另作说明,否则,芳基的每个独立地任选被取代,即,未取代(“未取代的芳基”)或被一个或多个取代基取代(“取代的芳基”)。在一些实施方案中,芳基是未取代的C6-14芳基。在一些实施方案中,芳基是取代的C6-14芳基。在一些实施方案中,芳基被一个或多个选自卤素、C1-C8烷基、C1-C8卤代烷基、氰基、羟基、C1-C8烷氧基和氨基的基团取代。取代的芳基的代表性的例子包括下列:其中,R56和R57中的一个可以是氢,R56和R57中的至少一个各自独立地选自:C1-C8烷基、C1-C8卤代烷基、4-10元杂环基、烷酰基、C1-C8烷氧基、杂芳氧基、烷基氨基、芳氨基、杂芳基氨基、NR58COR59、NR58SOR59、NR58SO2R59、COO烷基、COO芳基、CONR58R59、CONR58OR59、NR58R59、SO2NR58R59、S-烷基、SO烷基、SO2烷基、S芳基、SO芳基、SO2芳基;或R56和R57可以结合形成5至8个原子的环(饱和或不饱和),其任选含有一个或多个选自N、O或S的杂原子。R60和R61独立地是氢、C1-C8烷基、C1-C4卤代烷基、C3-C10环烷基、4-10元杂环基、C6-C10芳基、取代的C6-C10芳基、5-10元杂芳基或取代的5-10元杂芳基。具有稠合的杂环基的其它代表性的芳基包括下列:其中,每个W选自C(R66)2、NR66、O和S;每个Y选自羰基、NR66、O和S;R66独立地是氢、C1-C8烷基、C3-C10环烷基、4-10元杂环基、C6-C10芳基和5-10元杂芳基。“稠合的芳基”是指两个环碳与第二个芳基或杂芳基环共用或与碳环基或杂环基环共用的芳基。“芳烷基”是本文所定义的烷基和芳基的子集,是指被任选取代的芳基取代的任选取代的烷基。“杂芳基”是指具有提供在芳族环系中的环碳原子和1-4个环杂原子的5-10元单环或双环的4n+2芳族环体系(例如,具有以环状排列共享的6或10个π电子)的基团,其中每个杂原子独立地选自氮、氧和硫(“5-10元杂芳基”)。在含有一个或多个氮原子的杂芳基中,只要化合价允许,连接点可以是碳或氮原子。杂芳基双环系统在一个或两个环中可以包括一个或多个杂原子。“杂芳基”包括其中上述杂芳基环与一个或多个碳环基或杂环基稠合的环系,其中,连接点在杂芳基环上,且在这样的情况中,环成员的数目继续表示杂芳基环系中环成员的数目。“杂芳基”还包括其中上述杂芳基环与一个或多个芳基稠合的环系,其中,连接点在芳基或杂芳基环上,且在这样的情况中,环成员的数目表示稠合(芳基/杂芳基)环系中环成员的数目。其中一个环不包含杂原子的双环杂芳基(例如,吲哚基、喹啉基、咔唑基等),连接点可在任一环上,即,带有杂原子的环(例如,2-吲哚基)或不包含杂原子的环(例如,5-吲哚基)上。在一些实施方案中,杂芳基是具有环碳原子和1-4个环杂原子(存在于芳香环系中)的5-10元芳香环系,其中,每个杂原子独立地选自氮、氧和硫(“5-10元杂芳基”)。在一些实施方案中,杂芳基是具有环碳原子和1-4个环杂原子(存在于芳香环系中)的5-8元芳香环系,其中,每个杂原子独立地选自氮、氧和硫(“5-8元杂芳基”)。在一些实施方案中,杂芳基是具有环碳原子和1-4个环杂原子(存在于芳香环系中)的5-6元芳香环系,其中,每个杂原子独立地选自氮、氧和硫(“5-6元杂芳基”)。在一些实施方案中,5-6元杂芳基具有1-3个选自氮、氧和硫的环杂原子。在一些实施方案中,5-6元杂芳基具有1-2个选自氮、氧和硫的环杂原子。在一些实施方案中,5-6元杂芳基具有1个选自氮、氧和硫的环杂原子。除非另作说明,否则,杂芳基的每个独立地任选被取代的,即,未取代(“未取代的杂芳基”)或被一个或多个取代基取代(“取代的杂芳基”)。在一些实施方案中,杂芳基是未取代的5-14元杂芳基。在一些实施方案中,杂芳基是取代的5-14元杂芳基。示例性的含有一个杂原子的5元杂芳基包括但不限于:吡咯基、呋喃基和噻吩基。示例性的含有两个杂原子的5元杂芳基包括但不限于:咪唑基、吡唑基、噁唑基、异噁唑基、噻唑基和异噻唑基。示例性的含有三个杂原子的5元杂芳基包括但不限于:三唑基、噁二唑基和噻二唑基。示例性的含有四个杂原子的5元杂芳基包括但不限于:四唑基。示例性的含有一个杂原子的6元杂芳基包括但不限于:吡啶基。示例性的含有两个杂原子的6元杂芳基包括但不限于:哒嗪基、嘧啶基和吡嗪基。示例性的含有三个或四个杂原子的6元杂芳基分别包括但不限于:三嗪基和四嗪基。示例性的含有一个杂原子的7元杂芳基包括但不限于:氮杂环庚三烯基、氧杂环庚三烯基和硫杂环庚三烯基。示例性的5,6-双环杂芳基包括但不限于:吲哚基、异吲哚基、吲唑基、苯并三唑基、苯并噻吩基、异苯并噻吩基、苯并呋喃基、苯并异呋喃基、苯并咪唑基、苯并噁唑基、苯并异噁唑基、苯并噁二唑基、苯并噻唑基、苯并异噻唑基、苯并噻二唑基、茚嗪基和嘌呤基。示例性的6,6-双环杂芳基包括但不限于:萘啶基、喋啶基、喹啉基、异喹啉基、噌琳基、喹喔啉基、酞嗪基和喹唑啉基。代表性的杂芳基的例子包括下列:其中,每个Y选自羰基、N、NR65、O和S;R65独立地是氢、C1-C8烷基、C3-C10环烷基、4-10元杂环基、C6-C10芳基和5-10元杂芳基。“杂芳烷基”为本文所定义的烷基和杂芳基的子集,且是指被任选取代的杂芳基取代的任选取代的烷基。“碳环基”或“碳环”是指在非芳香环系中具有3至10个环碳原子和零个杂原子的非芳香环烃基团(“C3-10碳环基”)。在一些实施方案中,碳环基具有3至8个环碳原子(“C3-8碳环基”)。在一些实施方案中,碳环基具有3至6个环碳原子(“C3-6碳环基”)。在一些实施方案中,碳环基具有3至6个环碳原子(“C3-6碳环基”)。在一些实施方案中,碳环基具有5至10个环碳原子(“C5-10碳环基”)。示例性的C3-6碳环基包括但不限于:环丙基(C3)、环丙烯基(C3)、环丁基(C4)、环丁烯基(C4)、环戊基(C5)、环戊烯基(C5)、环己基(C6)、环己烯基(C6)、环已二烯基(C6),等。示例性的C3-8碳环基包括但不限于:上述C3-6碳环基,以及环庚基(C7)、环庚烯基(C7)、环庚二烯基(C7)、环庚三烯基(C7)、环辛基(C8)、环辛烯基(C8)、二环[2.2.1]庚基(C7)、二环[2.2.2]辛基(C8),等。示例性的C3-10碳环基包括但不限于:上述C3-8碳环基,以及环壬基(C9)、环壬烯基(C9)、环癸基(C10)、环癸烯基(C10)、八氢-1H-茚基(C9)、十氢萘基(C10)、螺[4.5]癸基(C10),等。正如前述实例所说明的那样,在一些实施方案中,碳环基为单环(“单环碳环基”)或包含稠环体系、桥环体系或螺环体系的碳环基,例如双环体系(“双环碳环基”),且可为饱和的或者可为部分不饱和的碳环基。“碳环基”还包括其中上述碳环基环与一个或多个芳基或杂芳基稠合的环体系,其中,连接点在碳环基环上,且在这样的情况中,碳的数目继续表示碳环体系中的碳的数目。除非另作说明,否则,碳环基的每个独立地为任选取代的,即,未取代的(“未取代的碳环基”)或被一个或多个取代基取代(“取代的碳环基”)。在一些实施方案中,碳环基是未取代的C3-10碳环基。在一些实施方案中,碳环基是取代的C3-10碳环基。在一些实施方案中,“碳环基”是具有3至10个环碳原子的单环的饱和碳环基(“C3-10环烷基”)。在一些实施方案中,环烷基具有3至8个环碳原子(“C3-8环烷基”)。在一些实施方案中,环烷基具有3至6个环碳原子(“C3-6环烷基”)。在一些实施方案中,环烷基具有5至6个环碳原子(“C5-6环烷基”)。在一些实施方案中,环烷基具有5至10个环碳原子(“C5-10环烷基”)。C5-6环烷基的例子包括环戊基(C5)和环己基(C6)。C3-6环烷基的例子包括上述C5-6环烷基,以及环丙基(C3)和环丁基(C4)。C3-8环烷基的例子包括上述C3-6环烷基,以及环庚基(C7)和环辛基(C8)。除非另作说明,否则,环烷基的每种情况独立地是未取代的(“未取代的环烷基”)或被一个或多个取代基取代(“取代的环烷基”)。在一些实施方案中,环烷基是未取代的C3-10环烷基。在一些实施方案中,环烷基是取代的C3-10环烷基。“杂环基”或“杂环”是指具有环碳原子和1至4个环杂原子的3至10元非芳香环系的原子团,其中,每个杂原子独立地选自氮、氧、硫、硼、磷和硅(“3-10元杂环基”)。在包含一个或多个氮原子的杂环基中,只要化合价允许,连接点可为碳或氮原子。杂环基可为单环的(“单环杂环基”)或稠环体系、桥环体系或螺环体系,例如双环体系(“双环杂环基”),且可为饱和的或者可为部分不饱和的杂环基。杂环基双环系统在一个或两个环中可以包括一个或多个杂原子。“杂环基”还包括其中上述杂环基环与一个或多个碳环基稠合的环体系,其中,连接点在碳环基或杂环基环上,或其中上述杂环基环与一个或多个芳基或杂芳基稠合的环体系,其中,连接点在杂环基环上,且在这样的情况下,环成员的数目继续表示在杂环基环体系中环成员的数目。除非另作说明,否则,杂环基的每个独立地为任选地取代的,即,未取代的(“未取代的杂环基”)或被一个或多个取代基取代(“取代的杂环基”)。在一些实施方案中,杂环基是未取代的3-10元杂环基。在一些实施方案中,杂环基是取代的3-10元杂环基。在一些实施方式中,杂环基为具有环碳原子和1-4个环杂原子的5-10元非芳香环系,其中,每个杂原子独立地选自氮、氧、硫、硼、磷和硅(“5-10元杂环基”)。在一些实施方式中,杂环基为具有环碳原子和1-4个环杂原子的5-8元非芳香环系,其中,每个杂原子独立地选自氮、氧和硫(“5-8元杂环基”)。在一些实施方式中,杂环基为具有环碳原子和1-4个环杂原子的5-6元非芳香环系,其中,每个杂原子独立地选自氮、氧和硫(“5-6元杂环基”)。在一些实施方案中,5-6元杂环基具有1-3个选自氮、氧和硫的环杂原子。在一些实施方案中,5-6元杂环基具有1-2个选自氮、氧和硫的环杂原子。在一些实施方案中,5-6元杂环基具有一个选自氮、氧和硫的环杂原子。示例性的包含一个杂原子的3元杂环基包括但不限于:氮杂环丙烷基、氧杂环丙烷基、硫杂环丙烷基(thiorenyl)。示例性的含有一个杂原子的4元杂环基包括但不限于:氮杂环丁烷基、氧杂环丁烷基和硫杂环丁烷基。示例性的含有一个杂原子的5元杂环基包括但不限于:四氢呋喃基、二氢呋喃基、四氢噻吩基、二氢噻吩基、吡咯烷基、二氢吡咯基和吡咯基-2,5-二酮。示例性的包含两个杂原子的5元杂环基包括但不限于:二氧杂环戊烷基、氧硫杂环戊烷基(oxasulfuranyl)、二硫杂环戊烷基(disulfuranyl)和噁唑烷-2-酮。示例性的包含三个杂原子的5元杂环基包括但不限于:三唑啉基、噁二唑啉基和噻二唑啉基。示例性的包含一个杂原子的6元杂环基包括但不限于:哌啶基、四氢吡喃基、二氢吡啶基和硫杂环己烷基(thianyl)。示例性的包含两个杂原子的6元杂环基包括但不限于:哌嗪基、吗啉基、二硫杂环己烷基、二噁烷基。示例性的包含三个杂原子的6元杂环基包括但不限于:六氢三嗪基(triazinanyl)。示例性的含有一个杂原子的7元杂环基包括但不限于:氮杂环庚烷基、氧杂环庚烷基和硫杂环庚烷基。示例性的包含一个杂原子的8元杂环基包括但不限于:氮杂环辛烷基、氧杂环辛烷基和硫杂环辛烷基。示例性的与C6芳基环稠合的5元杂环基(在本文中也称作5,6-双环杂环)包括但不限于:二氢吲哚基、异二氢吲哚基、二氢苯并呋喃基、二氢苯并噻吩基、苯并噁唑啉酮基,等。示例性的与芳基环稠合的6元杂环基(本文还指的是6,6-双环杂环)包括但不限于:四氢喹啉基、四氢异喹啉基,等。杂环基的具体实例示于以下说明性的实例中:其中,每个W选自CR67、C(R67)2、NR67、O和S;每个Y选自NR67、O和S;R67独立地是氢、C1-C8烷基、C3-C10环烷基、4-10元杂环基、C6-C10芳基、5-10元杂芳基。这些杂环基环可以任选被一个或多个选自下列的基团取代:酰基、酰氨基、酰氧基、烷氧基、烷氧羰基、烷氧基羰基氨基、氨基、取代的氨基、氨基羰基(氨基甲酰基或酰胺基)、氨基羰基氨基、氨基磺酰基、磺酰氨基、芳基、芳氧基、叠氮基、羧基、氰基、环烷基、卤素、羟基、酮基、硝基、硫醇、-S-烷基、-S-芳基、-S(O)-烷基、-S(O)-芳基、-S(O)2-烷基和-S(O)2-芳基。取代基包括羰基或硫代羰基,例如,提供内酰胺和脲衍生物。“杂”当用于描述化合物或存在于化合物上的基团时,是指所述化合物或基团中的一个或多个碳原子已被氮、氧或硫杂原子替代。杂可应用于上述任何烃基:例如,烷基,例如,杂烷基;环烷基,例如,杂环基;芳基,例如,杂芳基;环烯基,例如,环杂烯基等;其具有1至5个杂原子,尤其是1至3个杂原子。“酰基”是指基团-C(O)R20,其中,R20为氢、本文所定义的取代或未取代的烷基、取代或未取代的烯基、取代或未取代的炔基、取代或未取代的碳环基、取代或未取代的杂环基、取代或未取代的芳基、或取代或未取代的杂芳基。“烷酰基”是其中的R20是非氢基团的酰基。代表性的酰基包括但不限于:甲酰基(-CHO)、乙酰基(-C(=O)CH3)、环己基羰基、环己基甲基羰基、苯甲酰基(-C(=O)Ph)、苄基羰基(-C(=O)CH2Ph)、-C(O)-C1-C8烷基、-C(O)-(CH2)t(C6-C10芳基)、-C(O)-(CH2)t(5-10元杂芳基)、-C(O)-(CH2)t(C3-C10环烷基)和-C(O)-(CH2)t(4-10元杂环基),其中t为0至4的整数。在一些实施方案中,R21为被卤素或羟基取代的C1-C8烷基;或C3-C10环烷基、4-10元杂环基、C6-C10芳基、芳烷基、5-10元杂芳基或杂芳烷基取代,所述取代基各自被未取代的C1-C4烷基、卤素、未取代的C1-C4烷氧基、未取代的C1-C4卤代烷基、未取代的C1-C4羟基烷基或未取代的C1-C4卤代烷氧基或羟基取代。“酰氨基”是指基团-NR22C(O)R23,其中R22和R23的每个独立地为氢、本文所定义的取代或未取代的烷基、取代或未取代的烯基、取代或未取代的炔基、取代或未取代的碳环基、取代或未取代的杂环基、取代或未取代的芳基、或取代或未取代的杂芳基,或者R22为氨基保护基团。示例性的“酰氨基”包括但不限于:甲酰氨基、乙酰氨基、环己基羰基氨基、环己基甲基-羰基氨基、苯甲酰氨基和苄基羰基氨基。具体的示例性的“酰氨基”为-NR24C(O)-C1-C8烷基、-NR24C(O)-(CH2)t(C6-C10芳基)、-NR24C(O)-(CH2)t(5-10元杂芳基)、-NR24C(O)-(CH2)t(C3-C10环烷基)和-NR24C(O)-(CH2)t(4-10元杂环基),其中t为0至4的整数,且每个R24独立地代表H或C1-C8烷基。在一些实施方案中,R25为H、被卤素或羟基取代的C1-C8烷基;C3-C10环烷基、4-10元杂环基、C6-C10芳基、芳烷基、5-10元杂芳基或杂芳烷基,其各自被未取代的C1-C4烷基、卤素、未取代的C1-C4烷氧基、未取代的C1-C4卤代烷基、未取代的C1-C4羟基烷基或未取代的C1-C4卤代烷氧基或羟基取代;R26为H、被卤素或羟基取代的C1-C8烷基;C3-C10环烷基、4-10元杂环基、C6-C10芳基、芳烷基、5-10元杂芳基或杂芳烷基,其各自被未取代的C1-C4烷基、卤素、未取代的C1-C4烷氧基、未取代的C1-C4卤代烷基、未取代的C1-C4羟基烷基或未取代的C1-C4卤代烷氧基或羟基取代;条件是R25和R26中的至少一个不是H。“酰氧基”是指基团-OC(O)R27,其中,R27为氢、本文所定义的取代或未取代的烷基、取代或未取代的烯基、取代或未取代的炔基、取代或未取代的碳环基、取代或未取代的杂环基、取代或未取代的芳基或取代或未取代的杂芳基。代表性的实例包括但不限于:甲酰基、乙酰基、环己基羰基、环己基甲基羰基、苯甲酰基和苄基羰基。在一些实施方案中,R28为被卤素或羟基取代的C1-C8烷基;C3-C10环烷基、4-10元杂环基、C6-C10芳基、芳烷基、5-10元杂芳基或杂芳烷基,其各自被未取代的C1-C4烷基、卤素、未取代的C1-C4烷氧基、未取代的C1-C4卤代烷基、未取代的C1-C4羟基烷基或未取代的C1-C4卤代烷氧基或羟基取代。“烷氧基”是指基团-OR29,其中,R29为取代或未取代的烷基、取代或未取代的烯基、取代或未取代的炔基、取代或未取代的碳环基、取代或未取代的杂环基、取代或未取代的芳基或取代或未取代的杂芳基。具体烷氧基是甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、叔丁氧基、仲丁氧基、正戊氧基、正己氧基和1,2-二甲基丁氧基。具体烷氧基是低级烷氧基,即,具有1至6个碳原子。进一步的具体烷氧基具有1-4个碳原子。在一些实施方案中,R29为具有选自如下的1个或多个取代基的基团,例如,1至5个取代基,尤其是1至3个取代基,特别是1个取代基:氨基、取代的氨基、C6-C10芳基、芳氧基、羧基、氰基、C3-C10环烷基、4-10元杂环基、卤素、5-10元杂芳基、羟基、硝基、硫代烷氧基、硫代芳氧基、硫醇基、烷基-S(O)-、芳基-S(O)-、烷基-S(O)2-和芳基-S(O)2-。示例性的“取代的烷氧基”包括但不限于:-O-(CH2)t(C6-C10芳基)、-O-(CH2)t(5-10元杂芳基)、-O-(CH2)t(C3-C10环烷基)和-O-(CH2)t(4-10元杂环基),其中t为0至4的整数,且存在的任何芳基、杂芳基、环烷基或杂环基本身可被未取代的C1-C4烷基、卤素、未取代的C1-C4烷氧基、未取代的C1-C4卤代烷基、未取代的C1-C4羟基烷基、或未取代的C1-C4卤代烷氧基或羟基取代。具体的示例性的“取代的烷氧基”为-OCF3、-OCH2CF3、-OCH2Ph、-OCH2-环丙基、-OCH2CH2OH和-OCH2CH2NMe2。“氨基”是指基团-NH2。“取代的氨基”是指式-N(R38)2的氨基,其中,R38为氢、取代或未取代的烷基、取代或未取代的烯基、取代或未取代的炔基、取代或未取代的碳环基、取代或未取代的杂环基、取代或未取代的芳基、取代或未取代的杂芳基、或氨基保护基团,其中,至少一个R38不是氢。在一些实施方案中,每个R38独立地选自:氢、C1-C8烷基、C3-C8烯基、C3-C8炔基、C6-C10芳基、5-10元杂芳基、4-10元杂环基或C3-C10环烷基;或被卤素或羟基取代的C1-C8烷基;被卤素或羟基取代的C3-C8烯基;被卤素或羟基取代的C3-C8炔基,或-(CH2)t(C6-C10芳基)、-(CH2)t(5-10元杂芳基)、-(CH2)t(C3-C10环烷基)或-(CH2)t(4-10元杂环基),其中,t为0-8的整数,其各自被未取代的C1-C4烷基、卤素、未取代的C1-C4烷氧基、未取代的C1-C4卤代烷基、未取代的C1-C4羟基烷基或未取代的C1-C4卤代烷氧基或羟基取代;或者两个R38基团结合形成亚烷基。示例性的“取代的氨基”包括但不限于:-NR39-C1-C8烷基、-NR39-(CH2)t(C6-C10芳基)、-NR39-(CH2)t(5-10元杂芳基)、-NR39-(CH2)t(C3-C10环烷基)和-NR39-(CH2)t(4-10元杂环基),其中,t为0至4的整数,例如,1或2,每个R39独立地代表H或C1-C8烷基;存在的任何烷基本身可被卤素、取代或未取代的氨基或羟基取代;存在的任何芳基、杂芳基、环烷基或杂环基本身可被未取代的C1-C4烷基、卤素、未取代的C1-C4烷氧基、未取代的C1-C4卤代烷基、未取代的C1-C4羟基烷基或未取代的C1-C4卤代烷氧基或羟基取代。为了避免疑惑,术语“取代的氨基”包括基团烷基氨基、取代的烷基氨基、烷基芳基氨基、取代的烷基芳基氨基、芳基氨基、取代的芳基氨基、二烷基氨基和取代的二烷基氨基,如下面所定义。取代的氨基包括单取代的氨基和二取代的氨基。“叠氮基”是指基团-N3。“氨基甲酰基”或“酰胺基”是指基团-C(O)NH2。“取代的氨基甲酰基”或“取代的酰氨基”是指基团-C(O)N(R62)2,其中,每个R62独立地为氢、取代或未取代的烷基、取代或未取代的烯基、取代或未取代的炔基、取代或未取代的碳环基、取代或未取代的杂环基、取代或未取代的芳基、取代或未取代的杂芳基或氨基保护基团,其中,至少一个R62不是氢。在一些实施方案中,R62选自H、C1-C8烷基、C3-C10环烷基、4-10元杂环基、C6-C10芳基、芳烷基、5-10元杂芳基和杂芳烷基;或被卤素或羟基取代的C1-C8烷基;或C3-C10环烷基、4-10元杂环基、C6-C10芳基、芳烷基、5-10元杂芳基、或杂芳烷基,其各自被未取代的C1-C4烷基、卤素、未取代的C1-C4烷氧基、未取代的C1-C4卤代烷基、未取代的C1-C4羟基烷基或未取代的C1-C4卤代烷氧基或羟基取代;条件是,至少一个R62不是H。示例性的“取代的氨基甲酰基”包括但不限于:-C(O)NR64-C1-C8烷基、-C(O)NR64-(CH2)t(C6-C10芳基)、-C(O)N64-(CH2)t(5-10元杂芳基)、-C(O)NR64-(CH2)t(C3-C10环烷基)和-C(O)NR64-(CH2)t(4-10元杂环基),其中,t为0-4的整数,每个R64独立地代表H或C1-C8烷基,且存在的任何芳基、杂芳基、环烷基或杂环基本身可被未取代的C1-C4烷基、卤素、未取代的C1-C4烷氧基、未取代的C1-C4卤代烷基、未取代的C1-C4羟基烷基或未取代的C1-C4卤代烷氧基或羟基取代。“羧基”是指基团-C(O)OH。“氰基”是指基团-CN。“卤代”或“卤素”是指氟(F)、氯(Cl)、溴(Br)和碘(I)。在一些实施方案中,卤素基团是氟或氯。“羟基”是指基团-OH。“硝基”是指基团-NO2。“环烷基烷基”是指其中烷基被环烷基取代的烷基。典型的环烷基烷基包括但不限于:环丙基甲基、环丁基甲基、环戊基甲基、环己基甲基、环庚基甲基、环辛基甲基、环丙基乙基、环丁基乙基、环戊基乙基、环己基乙基、环庚基乙基和环辛基乙基,等。“杂环烷基”是指其中烷基被杂环基取代的烷基。典型的杂环基烷基包括但不限于:吡咯烷基甲基、哌啶基甲基、哌嗪基甲基、吗啉基甲基、吡咯烷基乙基、哌啶基乙基、哌嗪基乙基、吗啉基乙基,等。“环烯基”是指具有3至10个碳原子并具有单环或多个稠合环(包括稠环体系和桥环体系)且具有至少一个(特别是1至2个)烯属不饱和位点的取代或未取代的碳环基。这种环烯基包括,例如,单环结构,例如,环己烯基、环戊烯基、环丙烯基,等。“稠合环烯基”是指其两个环碳原子与第二个脂族或芳族环共用且使其烯属不饱和定位成赋予环烯基环以芳香性的环烯基。“亚乙基”是指取代或未取代的-(C-C)-。“乙烯基”是指取代或未取代的-(C=C)-。“乙炔基”是指-(C≡C)-。“含氮杂环基”是指包含至少一个氮原子的4至7元非芳香环基团,例如但但不限于:吗啉、哌啶(例如,2-哌啶基、3-哌啶基和4-哌啶基)、吡咯烷(例如,2-吡咯烷基和3-吡咯烷基)、氮杂环丁烷、吡咯烷酮、咪唑啉、咪唑烷酮、2-吡唑啉、吡唑烷、哌嗪和N-烷基哌嗪,例如,N-甲基哌嗪。具体实例包括氮杂环丁烷、哌啶酮和哌嗪酮。“硫酮基”是指基团=S。本文定义的烷基、烯基、炔基、碳环基、杂环基、芳基和杂芳基为任选取代的基团(例如,“取代的”或“未取代的”烷基、“取代的”或“未取代的”烯基、“取代的”或“未取代的”炔基、“取代的”或“未取代的”碳环基、“取代的”或“未取代的”杂环基、“取代的”或“未取代的”芳基或者“取代的”或“未取代的”杂芳基)。通常,术语“取代的”,不论前面是否有术语“任选”,是指存在于基团(例如,碳或氮原子)上的至少一个氢被可允许的取代基取代,例如,在取代时产生稳定的化合物的取代基,例如,不自发地进行转变(例如通过重排、环化、消除或其它反应)的化合物。除非另外说明,否则,“取代的”基团在所述基团的一个或多个可取代的位置处具有取代基,且当在任何给定结构中的一个以上的位置被取代时,在每个位置处的取代基是相同或不同的。术语“取代的”包括用有机化合物的所有可允许的取代基(导致形成稳定化合物的本文描述的任何取代基)进行的取代。对于本发明,杂原子例如氮可具有氢取代基和/或本文描述的任何合适的取代基,其满足杂原子的化合价且导致形成稳定的部分。示例性的碳原子取代基包括但不限于:卤素、-CN、-NO2、-N3、-SO2H、-SO3H、-OH、-ORaa、-ON(Rbb)2、-N(Rbb)2、-N(Rbb)3+X-、-N(ORcc)Rbb、-SH、-SRaa、-SSRcc、-C(=O)Raa、-CO2H、-CHO、-C(ORcc)2、-CO2Raa、-OC(=O)Raa、-OCO2Raa、-C(=O)N(Rbb)2、-OC(=O)N(Rbb)2、-NRbbC(=O)Raa、-NRbbCO2Raa、-NRbbC(=O)N(Rbb)2、-C(=NRbb)Raa、-C(=NRbb)ORaa、-OC(=NRbb)Raa、-OC(=NRbb)ORaa、-C(=NRbb)N(Rbb)2、-OC(=NRbb)N(Rbb)2、-NRbbC(=NRbb)N(Rbb)2、-C(=O)NRbbSO2Raa、-NRbbSO2Raa、-SO2N(Rbb)2、-SO2Raa、-SO2ORaa、-OSO2Raa、-S(=O)Raa、-OS(=O)Raa、-Si(Raa)3、-OSi(Raa)3、-C(=S)N(Rbb)2、-C(=O)SRaa、-C(=S)SRaa、-S(O)Raa、例如-SC(=S)SRaa、-SC(=O)SRaa、-OC(=O)SRaa、-SC(=O)ORaa、-SC(=O)Raa、-P(=O)2Raa、-OP(=O)2Raa、-P(=O)(Raa)2、-OP(=O)(Raa)2、-OP(=O)(ORcc)2、-P(=O)2N(Rbb)2、-OP(=O)2N(Rbb)2、-P(=O)(NRbb)2、-OP(=O)(NRbb)2、-NRbbP(=O)(ORcc)2、-NRbbP(=O)(NRbb)2、-P(Rcc)2、-P(Rcc)3、-OP(Rcc)2、-OP(Rcc)3、-B(Raa)2、-B(ORcc)2、-BRaa(ORcc)、C1-10烷基、C1-10全卤代烷基、C2-10烯基、C2-10炔基、C3-10碳环基、3-14元杂环基、C6-14芳基和5-14元杂芳基,其中,每个烷基、烯基、炔基、碳环基、杂环基、芳基和杂芳基独立地被0、1、2、3、4或5个Rdd基团取代;或者在碳原子上的两个偕氢(geminalhydrogen)被基团=O、=S、=NN(Rbb)2、=NNRbbC(=O)Raa、=NNRbbC(=O)ORaa、=NNRbbS(=O)2Raa、=NRbb或=NORcc取代;Raa的每个独立地选自C1-10烷基、C1-10全卤代烷基、C2-10烯基、C2-10炔基、C3-10碳环基、3-14元杂环基、C6-14芳基和5-14元杂芳基,或者两个Raa基团结合以形成3-14元杂环基或5-14元杂芳基环,其中,每个烷基、烯基、炔基、碳环基、杂环基、芳基和杂芳基独立地被0、1、2、3、4或5个Rdd基团取代;Rbb的每个独立地选自:氢、-OH、-ORaa、-N(Rcc)2、-CN、-C(=O)Raa、-C(=O)N(Rcc)2、-CO2Raa、-SO2Raa、-C(=NRcc)ORaa、-C(=NRcc)N(Rcc)2、-SO2N(Rcc)2、-SO2Rcc、-SO2ORcc、-SORaa、-C(=S)N(Rcc)2、-C(=O)SRcc、-C(=S)SRcc、-P(=O)2Raa、-P(=O)(Raa)2、-P(=O)2N(Rcc)2、-P(=O)(NRcc)2、C1-10烷基、C1-10全卤代烷基、C2-10烯基、C2-10炔基、C3-10碳环基、3-14元杂环基、C6-14芳基和5-14元杂芳基,或者两个Rbb基团结合以形成3-14元杂环基或5-14元杂芳基环,其中,每个烷基、烯基、炔基、碳环基、杂环基、芳基和杂芳基独立地被0、1、2、3、4或5个Rdd基团取代;Rcc的每个独立地选自氢、C1-10烷基、C1-10全卤代烷基、C2-10烯基、C2-10炔基、C3-10碳环基、3-14元杂环基、C6-14芳基和5-14元杂芳基,或者两个Rcc基团结合以形成3-14元杂环基或5-14元杂芳基环,其中,每个烷基、烯基、炔基、碳环基、杂环基、芳基和杂芳基独立地被0、1、2、3、4或5个Rdd基团取代;Rdd的每个独立地选自:卤素、-CN、-NO2、-N3、-SO2H、-SO3H、-OH、-ORee、-ON(Rff)2、-N(Rff)2,、-N(Rff)3+X-、-N(ORee)Rff、-SH、-SRee、-SSRee、-C(=O)Ree、-CO2H、-CO2Ree、-OC(=O)Ree、-OCO2Ree、-C(=O)N(Rff)2、-OC(=O)N(Rff)2、-NRffC(=O)Ree、-NRffCO2Ree、-NRffC(=O)N(Rff)2、-C(=NRff)ORee、-OC(=NRff)Ree、-OC(=NRff)ORee、-C(=NRff)N(Rff)2、-OC(=NRff)N(Rff)2、-NRffC(=NRff)N(Rff)2、-NRffSO2Ree、-SO2N(Rff)2、-SO2Ree、-SO2ORee、-OSO2Ree、-S(O)Ree,例如-S(=O)Ree、-Si(Ree)3、-OSi(Ree)3、-C(=S)N(Rff)2、-C(=O)SRee、-C(=S)SRee、-SC(=S)SRee、-P(=O)2Ree、-P(=O)(Ree)2、-OP(=O)(Ree)2、-OP(=O)(ORee)2、C1-6烷基、C1-6全卤代烷基、C2-6烯基、C2-6炔基、C3-10碳环基、3-10元杂环基、C6-10芳基、5-10元杂芳基,其中,每个烷基、烯基、炔基、碳环基、杂环基、芳基和杂芳基独立地被0、1、2、3、4或5个Rgg基团取代,或者两个偕Rdd取代基可结合以形成=O或=S;Ree的每个独立地选自C1-6烷基、C1-6全卤代烷基、C2-6烯基、C2-6炔基、C3-10碳环基、C6-10芳基、3-10元杂环基和3-10元杂芳基,其中,每个烷基、烯基、炔基、碳环基、杂环基、芳基和杂芳基独立地被0、1、2、3、4或5个Rgg基团取代;Rff的每个独立地选自氢、C1-6烷基、C1-6全卤代烷基、C2-6烯基、C2-6炔基、C3-10碳环基、3-10元杂环基、C6-10芳基和5-10元杂芳基,或者两个Rff基团结合形成3-14元杂环基或5-14元杂芳基环,其中,每个烷基、烯基、炔基、碳环基、杂环基、芳基和杂芳基独立地被0、1、2、3、4或5个Rgg基团取代;Rgg的每个独立地是:卤素、-CN、-NO2、-N3、-SO2H、-SO3H、-OH、-OC1-6烷基、-ON(C1-6烷基)2、-N(C1-6烷基)2、-N(C1-6烷基)3+X-、-NH(C1-6烷基)2+X-、-NH2(C1-6烷基)+X-、-NH3+X-、-N(OC1-6烷基)(C1-6烷基)、-N(OH)(C1-6烷基)、-NH(OH)、-SH、-SC1-6烷基、-SS(C1-6烷基)、-C(=O)(C1-6烷基)、-CO2H、-CO2(C1-6烷基)、-OC(=O)(C1-6烷基)、-OCO2(C1-6烷基)、-C(=O)NH2、-C(=O)N(C1-6烷基)2、-OC(=O)NH(C1-6烷基)、-NHC(=O)(C1-6烷基)、-N(C1-6烷基)C(=O)(C1-6烷基)、-NHCO2(C1-6烷基)、-NHC(=O)N(C1-6烷基)2、-NHC(=O)NH(C1-6烷基)、-NHC(=O)NH2、-C(=NH)O(C1-6烷基)、-OC(=NH)(C1-6烷基)、-OC(=NH)OC1-6烷基、-C(=NH)N(C1-6烷基)2、-C(=NH)NH(C1-6烷基)、-C(=NH)NH2、-OC(=NH)N(C1-6烷基)2、-OC(NH)NH(C1-6烷基)、-OC(NH)NH2、-NHC(NH)N(C1-6烷基)2、-NHC(=NH)NH2、-NHSO2(C1-6烷基)、-SO2N(C1-6烷基)2、-SO2NH(C1-6烷基)、-SO2NH2、-SO2C1-6烷基、-SO2OC1-6烷基、-OSO2C1-6烷基、-SOC1-6烷基、-Si(C1-6烷基)3、-OSi(C1-6烷基)3、-C(=S)N(C1-6烷基)2、C(=S)NH(C1-6烷基)、C(=S)NH2、-C(=O)S(C1-6烷基)、-C(=S)SC1-6烷基、-SC(=S)SC1-6烷基、-P(=O)2(C1-6烷基)、-P(=O)(C1-6烷基)2、-OP(=O)(C1-6烷基)2、-OP(=O)(OC1-6烷基)2、C1-6烷基、C1-6全卤代烷基、C2-6烯基、C2-6炔基、C3-10碳环基、C6-10芳基、3-10元杂环基、5-10元杂芳基;或者两个偕Rgg取代基可结合形成=O或=S;其中,X-为反离子。“反离子”或“阴离子型反离子”为带负电的基团,其与阳离子季氨基结合以保持电中性。示例性的反离子包括:卤根离子(例如,F-、Cl-、Br-、I-)、NO3-、ClO4-、OH-、H2PO4-、HSO4-、SO4-2、磺酸根离子(例如,甲磺酸根、三氟甲磺酸根、对甲苯磺酸根、苯磺酸根、10-樟脑磺酸根、萘-2-磺酸根、萘-1-磺酸-5-磺酸根、乙烷-1-磺酸-2-磺酸根等),以及羧酸根离子(例如,醋酸根(acetate)、乙酸根(ethanoate)、丙酸根、苯甲酸根、甘油酸根、乳酸根、酒石酸根、乙醇酸根,等)。只要化合价允许,氮原子可为取代或未取代的,且包括伯、仲、叔和季氮原子。示例性的氮原子取代基包括但不限于:氢、-OH、-ORaa、-N(Rcc)2、-CN、-C(=O)Raa、-C(=O)N(Rcc)2、-CO2Raa、-SO2Raa、-C(=NRbb)Raa、-C(=NRcc)ORaa、-C(=NRcc)N(Rcc)2、-SO2N(Rcc)2、-SO2Rcc、-SO2ORcc、-SORaa、-C(=S)N(Rcc)2、-C(=O)SRcc、-C(=S)SRcc、-P(=O)2Raa、-P(=O)(Raa)2、-P(=O)2N(Rcc)2、-P(=O)(NRcc)2、C1-10烷基、C1-10全卤代烷基、C2-10烯基、C2-10炔基、C3-10碳环基、3-14元杂环基、C6-14芳基和5-14元杂芳基,或者连接至氮原子的两个Rcc基团结合形成3-14元杂环基或5-14元杂芳基环,其中,每个烷基、烯基、炔基、碳环基、杂环基、芳基和杂芳基独立地被0、1、2、3、4或5个Rdd基团取代,且其中Raa、Rbb、Rcc和Rdd如上所述。在详细说明、实施例和权利要求中,更详细地描述这些及其它示例性的取代基。本发明不是意在以任何方式受到上面示例性的取代基限制。其它定义术语“可药用盐”是指,在可靠的医学判断范围内,适合与人和低等动物的组织接触而没有过度毒性、刺激性、变态反应等,并且与合理的益处/危险比例相称的那些盐。可药用盐在本领域是众所周知的。例如,Berge等人在J.PharmaceuticalSciences(1977)66:1-19中详细描述的可药用盐。本发明化合物的可药用盐包括衍生自合适无机和有机酸和碱的盐。可药用无毒酸加成盐的例子是氨基与无机酸形成的盐,例如盐酸、氢溴酸、磷酸、硫酸和高氯酸,或与有机酸形成的盐,例如乙酸、草酸、马来酸、酒石酸、枸橼酸、琥珀酸或丙二酸,或使用本领域使用的方法形成的盐,例如,离子交换方法。其它可药用盐包括:已二酸盐、海藻酸盐、抗坏血酸盐、天冬氨酸盐、苯磺酸盐、苯甲酸盐、重硫酸盐、硼酸盐、丁酸盐、樟脑酸盐、樟脑磺酸盐、柠檬酸盐、环戊丙酸盐、二葡糖酸盐、十二烷基硫酸盐、乙磺酸盐、甲酸盐、富马酸盐、葡萄糖酸盐、甘油磷酸盐、葡糖酸盐、半硫酸盐、庚酸盐、己酸盐、氢碘酸盐、2-羟基-乙磺酸盐、乳糖酸盐、乳酸盐、月桂酸盐、月桂基硫酸盐、苹果酸盐、马来酸盐、丙二酸盐、甲磺酸盐、2-萘磺酸盐、烟酸盐、硝酸盐、油酸盐、草酸盐、棕榈酸盐、双羟萘酸盐、果胶酯酸盐、过硫酸盐、3-苯丙酸盐、磷酸盐、苦味酸盐、特戊酸盐、丙酸盐、硬脂酸盐、琥珀酸盐、硫酸盐、酒石酸盐、硫氰酸盐、对甲苯磺酸盐、十一烷酸盐、戊酸盐,等。衍生自合适的碱的可药用盐包括碱金属、碱土金属、铵和N+(C1-4烷基)4盐。代表性的碱金属或碱土金属盐包括钠、锂、钾、钙、镁盐,等。如果合适的话,进一步的可药用盐包括使用反离子形成的无毒的铵盐、季铵盐和胺阳离子,反离子例如卤离子、氢氧根、羧酸根、硫酸根、磷酸根、硝酸根、低级烷基磺酸根和芳基磺酸根。施用的“受试者”包括但不限于:人(即,任何年龄组的男性或女性,例如,儿科受试者(例如,婴儿、儿童、青少年)或成人受试者(例如,年轻的成人、中年的成人或年长的成人))和/或非人的动物,例如,哺乳动物,例如,灵长类(例如,食蟹猴、恒河猴)、牛、猪、马、绵羊、山羊、啮齿动物、猫和/或狗。在一些实施方案中,受试者是人。在一些实施方案中,受试者是非人动物。本文可互换使用术语“人”、“患者”和“受试者”。疾病、障碍和病症在本文中可互换地使用。除非另作说明,否则,本文使用的术语“治疗”包括受试者患有具体疾病、障碍或病症时所发生的作用,它降低疾病、障碍或病症的严重程度,或延迟或减缓疾病、障碍或病症的发展(“治疗性治疗”),还包括受试者开始患有具体疾病、障碍或病症之前发生的作用(“预防性治疗”)。通常,化合物的“有效量”是指足以引起目标生物反应的量。正如本领域普通技术人员所理解的那样,本发明化合物的有效量可以根据下列因素而改变:例如,生物学目标、化合物的药物动力学、所治疗的疾病、施用模式以及受试者的年龄健康情况和症状。有效量包括治疗有效量和预防性治疗有效量。除非另作说明,否则,本文使用的化合物的“治疗有效量”是在治疗疾病、障碍或病症的过程中足以提供治疗益处的量,或使与疾病、障碍或病症有关的一或多种症状延迟或最小化。化合物的治疗有效量是指单独使用或与其它疗法联用的治疗剂的量,它在治疗疾病、障碍或病症的过程中提供治疗益处。术语“治疗有效量”可以包括改善总体治疗、降低或避免疾病或病症的症状或病因、或增强其它治疗剂的治疗效能的量。除非另作说明,否则,本文使用的化合物的“预防有效量”是足以预防疾病、障碍或病症的量,或足以预防与疾病、障碍或病症有关的一或多种症状的量,或防止疾病、障碍或病症复发的量。化合物的预防有效量是指单独使用或与其它药剂联用的治疗剂的量,它在预防疾病、障碍或病症的过程中提供预防益处。术语“预防有效量”可以包括改善总体预防的量,或增强其它预防药剂的预防效能的量。附图简述图1-22描绘了本文中描述的例示性化合物的代表性1HNMR谱。发明详述如本文中描述,本发明提供了式(I)的19-去甲C3,3-二取代的C21-三唑和C21-四唑神经活性类固醇或其药学可接受盐:其中:A选自下组:R1是C1-C6卤代烷基(CHF2,CH2F)或C1-C6烷基(例如,CH3,CH2CH3,CH2OCH3,CH2OCH2CH3);R2和R3独立选自:H,卤素(例如,F),C1-C6烷基(例如,CH3)或烷氧基(OCH3,OCH2CH3);R4是卤素(例如,Cl,F),氰基,硝基,-S(O)xRa,-NRbRc,C1-C6烷基(例如,CH3,CF3),C1-C6烷氧基,-C(O)Ra,-C(O)ORa,或-C(O)NRbRc;Ra是H或C1-C6烷基;每个Rb和Rc独立是H,-S(O)xRa,-C(O)Ra,C1-C6烷基,或C1-C6烷氧基,或Rb和Rc与它们附接的原子一起形成环(例如,Rb和Rc与它们附接的原子一起形成4-8元环,例如,杂环状环,例如,吗啉环,吡咯烷环,哌啶环);n是0至2的整数;并且x是0至2的整数。在一些实施方案中,当A是(A-1)或(A-2)时,则R1选自:-CHF2,CH2F,-CCl3,-CHCl2,CH2Cl,-CBr3,CHBr2,CH2Br,或C1-C6烷基;或者当A是(A-3)或(A-5)时,R1是CH3,-CH2F,-CH2OCH3,或–CHF2,并且n是0,则R2和R3的至少一个不是H。在一些实施方案中,当A是(A-1),(A-3),或(A-5),并且n是0时,则R2和R3的至少一个不是H。在一些实施方案中,当A是(A-1),(A-3),或(A-5),则R2和R3的至少一个不是H。在一些实施方案中,当A是(A-1)或(A-2),并且n是0时,则R1选自:CHF2,CH2F,-CCl3,-CHCl2,CH2Cl,-CBr3,CHBr2,CH2Br,或C1-C6烷基。在一些实施方案中,n是0或1。在一些实施方案中,n是0。在一些实施方案中,n是1。在一些实施方案中,式(I)的化合物选自式(Ia)的化合物:在一些实施方案中,式(I)的化合物选自式(Ib)的化合物:在一些实施方案中,式(I)的化合物选自式(II)的化合物:在一些实施方案中,n是1并且R4是卤素,氰基,-S(O)xRa,或C1-C6烷基。在一些实施方案中,R4是-CH3。在一些实施方案中,R4是氰基。在一些实施方案中,R4是-S(O)2CH3。在一些实施方案中,A选自下组:在一些实施方案中,R1是C1-C6烷基。在一些实施方案中,R1是-CH3。在一些实施方案中,R2和R3是H。在一些实施方案中,n是1并且R4是卤素,氰基,-S(O)xRa,或C1-C6烷基。在一些实施方案中,R4是-CH3。在一些实施方案中,R4是-C(O)ORa。在一些实施方案中,Ra是H。在一些实施方案中,Ra是C1-C6烷基。在一些实施方案中,Ra是–CH2CH3。在一些实施方案中,R4是-C(O)NRbRc。在一些实施方案中,Rb和Rc是H。在一些实施方案中,R4是氰基.在一些实施方案中,R4是-S(O)2CH3。在一些实施方案中,A选自下组:在一些实施方案中,化合物选自下组:在一方面,本文中提供了包含如本文中所述的化合物(例如,式(I)的化合物)或其药学可接受盐和药学可接受赋形剂的药物组合物。在一个方面,本发明提供用于在有此需要的受试者中治疗CNS相关病症的方法,其包括向受试者施用有效量的本文中所述的化合物(例如式(I)的化合物)或其药学可接受盐。在一些实施方案中,CNS相关病症是睡眠障碍,进食障碍,心境障碍,精神分裂症谱系障碍,惊厥性障碍,记忆和/或认知障碍,运动障碍,人格障碍,孤独症谱系障碍,疼痛,创伤性脑损伤,血管疾病,物质滥用障碍和/或戒断综合征或耳鸣。在一些实施方案中,CNS相关障碍是抑郁症(例如,产后抑郁)。在一些实施方案中,CNS相关病症是震颤(例如,特发性震颤)。在一些实施方案中,CNS相关病症是进食障碍(例如,神经性厌食,神经性贪食,贪吃症,恶病质)。在一些实施方案中,经口,皮下,静脉内或肌肉内施用所述化合物。在一些实施方案中,长期施用该化合物。在一个方面,本文提供了在受试者中诱导镇静和/或麻醉的方法,其包括向受试者施用有效量的式(I)的化合物。在一个方面,本文提供了用于治疗受试者中癫痫的方法,其包括向受试者施用有效量的式(I)的化合物。在一个方面,本文提供了用于治疗受试者中的癫痫的方法,所述方法包括向受试者施用有效量的式(I)的化合物。在一个方面,本文提供治疗受试者中癫痫持续状态(SE)的方法,所述方法包括向受试者施用有效量的式(I)的化合物。在一些实施方案中,癫痫持续状态是惊厥性癫痫持续状态(convulsivestatusepilepticus)(例如,早期癫痫持续状态(earlystatusepilepticus),建立的癫痫持续状态(establishedstatusepilepticus),难治性癫痫持续状态(refractorystatusepilepticus),超难治性癫痫持续状态(super-refractorystatusepilepticus))或非惊厥性癫痫持续状态,(例如,全身性癫痫持续状态(generalizedstatusepilepticus),复合部分型癫痫持续状态(complexpartialstatusepilepticus))。在一个方面,本文提供了用于治疗有此需要的受试者的病症(例如,如本文中所述的病症,例如,与GABA功能相关的病症)的方法,所述方法包括向所述受试者施用治疗有效量的化合物,其药学可接受的盐,或式(I)化合物之一的药物组合物。药物组合物在另一方面,本发明提供了药物组合物,其包含本发明的化合物(还称为“活性组分”)和可药用赋形剂。在一些实施方案中,所述药物组合物包含有效量的活性组分。在一些实施方案中,所述药物组合物包含治疗有效量的活性组分。在一些实施方案中,所述药物组合物包含预防有效量的活性组分。本发明提供的药物组合物可以通过许多途径施用,包括但不限于:口服(肠内)施用、肠胃外(注射)施用、直肠施用、透皮施用、皮内施用、鞘内施用、皮下(SC)施用、静脉内(IV)施用、肌内(IM)施用和鼻内施用。通常,施用有效量的本文所提供的化合物。按照有关情况,包括所治疗的病症、选择的施用途径、实际施用的化合物、个体患者的年龄、体重和响应、患者症状的严重程度,等,可以由医生确定实际上施用的化合物的量。当用于预防CNS病症发作时,对处于形成所述病症危险之中的受试者施用本文所提供的化合物,典型地基于医生的建议并在医生监督下施用,剂量水平如上所述。处于形成具体病症的风险之中的受试者,通常包括具有所述病症的家族史的受试者,或通过遗传试验或筛选确定尤其对形成所述病症敏感的那些受试者。还可以长期施用本文所提供的药物组合物(“长期施用”)。长期施用是指在长时间内施用化合物或其药物组合物,例如,3个月、6个月、1年、2年、3年、5年等,或者可无限期地,例如,在受试者的余生中持续施用。在一些实施方案中,长期施用意欲在长时间内,例如,在治疗窗内在血液中提供所述化合物的恒定水平。可以使用各种施用方法,进一步递送本发明的药物组合物。例如,在一些实施方案中,可以推注施用药物组合物,例如,以便使化合物在血液中的浓度提高至有效水平。推注剂量的配置取决于通过身体的活性组分的目标全身性水平,例如,肌内或皮下的推注剂量使活性组分缓慢释放,而直接递送至静脉的推注(例如,通过IV静脉滴注)能够更加快速地递送,使得活性组分在血液中的浓度快速升高至有效水平。在其它实施方案中,可以以持续输液形式施用药物组合物,例如,通过IV静脉滴注,从而在受试者身体中提供稳态浓度的活性组分。此外,在其它实施方案中,可以首先施用推注剂量的药物组合物,而后持续输液。口服组合物可以采用散装液体溶液或混悬剂或散装粉剂形式。然而,更通常,为了便于精确地配制,以单位剂量形式提供所述组合物。术语“单位剂型”是指适合作为人类患者及其它哺乳动物的单元剂量的物理离散单位,每个单位包含预定数量的、适于产生所需要的治疗效果的活性物质与合适药学赋形剂。典型的单位剂量形式包括液体组合物的预装填的、预先测量的安瓿或注射器,或者在固体组合物情况下的丸剂、片剂、胶囊剂等。在这种组合物中,所述化合物通常为较少的组分(约0.1至约50重量%,或优选约1至约40重量%),剩余部分为对于形成所需剂量形式有用的各种载体或赋形剂以及加工助剂。对于口服剂量,代表性的方案是,每天一个至五个口服剂量,尤其是两个至四个口服剂量,典型地是三个口服剂量。使用这些剂量给药模式,每个剂量提供大约0.01至大约20mg/kg的本发明提供的化合物,优选的剂量各自提供大约0.1至大约10mg/kg,尤其是大约1至大约5mg/kg。为了提供与使用注射剂量类似的血液水平,或比使用注射剂量更低的血液水平,通常选择透皮剂量,数量为大约0.01至大约20%重量,优选大约0.1至大约20%重量,优选大约0.1至大约10%重量,且更优选大约0.5至大约15%重量。从大约1至大约120小时,尤其是24至96小时,注射剂量水平在大约0.1mg/kg/小时至至少10mg/kg/小时的范围。为了获得足够的稳定状态水平,还可以施用大约0.1mg/kg至大约10mg/kg或更多的预载推注。对于40至80kg的人类患者来说,最大总剂量不能超过大约2g/天。适于口服施用的液体形式可包括合适的水性或非水载体以及缓冲剂、悬浮剂和分散剂、着色剂、调味剂,等。固体形式可包括,例如,任何下列组份,或具有类似性质的化合物:粘合剂,例如,微晶纤维素、黄蓍胶或明胶;赋形剂,例如,淀粉或乳糖,崩解剂,例如,褐藻酸、Primogel或玉米淀粉;润滑剂,例如,硬脂酸镁;助流剂,例如,胶体二氧化硅;甜味剂,例如,蔗糖或糖精;或调味剂,例如,薄荷、水杨酸甲酯或橙味调味剂。可注射的组合物典型地基于可注射用的无菌盐水或磷酸盐缓冲盐水,或本领域中已知的其它可注射的赋形剂。如前所述,在这种组合物中,活性化合物典型地为较少的组分,经常为约0.05至10%重量,剩余部分为可注射的赋形剂等。典型地将透皮组合物配制为含有活性组分的局部软膏剂或乳膏剂。当配制为软膏剂时,活性组分典型地与石蜡或可与水混溶的软膏基质组合。或者,活性组分可与例如水包油型乳膏基质一起配制为乳膏剂。这种透皮制剂是本领域中公知的,且通常包括用于提升活性组分或制剂的稳定的皮肤渗透的其它组份。所有这种已知的透皮制剂和组份包括在本发明提供的范围内。本发明提供的化合物还可通过透皮装置施用。因此,透皮施用可使用贮存器(reservoir)或多孔膜类型、或者多种固体基质的贴剂实现。用于口服施用、注射或局部施用的组合物的上述组份仅仅是代表性的。其它材料以及加工技术等阐述于Remington'sPharmaceuticalSciences,17thedition,1985,MackPublishingCompany,Easton,Pennsylvania的第8部分中,本文以引用的方式引入该文献。本发明的化合物还可以以持续释放形式施用,或从持续释放施用系统中施用。代表性的持续释放材料的描述可在Remington'sPharmaceuticalSciences中找到。本发明还涉及本发明化合物的可药用制剂。在一个实施方案中,所述制剂包含水。在另一个实施方案中,所述制剂包含环糊精衍生物。最常见的环糊精为分别由6、7和8个α-1,4-连接的葡萄糖单元组成的α-、β-和γ-环糊精,其在连接的糖部分上任选包括一个或多个取代基,其包括但不限于:甲基化的、羟基烷基化的、酰化的和磺烷基醚取代。在一些实施方案中,所述环糊精为磺烷基醚β-环糊精,例如,磺丁基醚β-环糊精,也称作参见,例如,U.S.5,376,645。在一些实施方案中,所述制剂包括六丙基-β-环糊精(例如,在水中,10-50%)。本发明还涉及本发明化合物的可药用酸加成盐。可用于制备可药用盐的酸为形成无毒的酸加成盐的酸,即,包含药理学上可接受的阴离子的盐,例如,盐酸盐、氢碘酸盐、氢溴酸盐、硝酸盐、硫酸盐、硫酸氢盐、磷酸盐、乙酸盐、乳酸盐、柠檬酸盐、酒石酸盐、琥珀酸盐、马来酸盐、富马酸盐、苯甲酸盐、对甲苯磺酸盐等。下列制剂实施例说明可根据本发明制备的代表性的药物组合物。然而,本发明不限于下列药物组合物。示例性的制剂1-片剂:可以将干粉形式的本发明化合物与干燥的凝胶粘合剂以约1:2的重量比混合。加入较少量的硬脂酸镁作为润滑剂。使该混合物在压片机中成型为240-270mg片剂(每个片剂含有80-90mg活性化合物)。示例性的制剂2-胶囊剂:可以将干粉形式的本发明化合物与淀粉稀释剂以约1:1的重量比混合。将该混合物填充到250mg胶囊中(每个胶囊含有125mg活性化合物)。示例性的制剂3-液体:可以将本发明的化合物(125mg)与蔗糖(1.75g)和黄原胶(4mg)混合,且可将得到的混合物共混,通过No.10筛目美国筛,然后与预先制备的微晶纤维素和羧甲基纤维素钠(11:89,50mg)的水溶液混合。将苯甲酸钠(10mg)、调味剂和着色剂用水稀释,并在搅拌下加入。然后,可以加入充足的水,得到5mL的总体积。示例性的制剂4-片剂:可以将干粉形式的本发明化合物与干燥的凝胶粘合剂以约1:2的重量比混合。加入较少量的硬脂酸镁作为润滑剂。使该混合物在压片机中成型为450-900mg片剂(150-300mg活性化合物)。示例性的制剂5-注射剂:可以将本发明的化合物溶解或悬浮在缓冲无菌盐水可注射的水性介质中,达到约5mg/mL的浓度。示例性的制剂6-片剂:可以将干粉形式的本发明化合物与干燥的凝胶粘合剂以约1:2的重量比混合。加入较少量的硬脂酸镁作为润滑剂。使该混合物在压片机中成型为90-150mg片剂(每个片剂含有30-50mg活性化合物)。示例性的制剂7-片剂:可以将干粉形式的本发明化合物与干燥的凝胶粘合剂以约1:2的重量比混合。加入较少量的硬脂酸镁作为润滑剂。使该混合物在压片机中成型为30-90mg片剂(每个片剂含有10-30mg活性化合物)。示例性的制剂8-片剂:可以将干粉形式的本发明化合物与干燥的凝胶粘合剂以约1:2的重量比混合。加入较少量的硬脂酸镁作为润滑剂。使该混合物在压片机中成型为0.3-30mg片剂(每个片剂含有0.1-10mg活性化合物)。示例性的制剂9-片剂:可以将干粉形式的本发明化合物与干燥的凝胶粘合剂以约1:2的重量比混合。加入较少量的硬脂酸镁作为润滑剂。使该混合物在压片机中成型为150-240mg片剂(每个片剂含有50-80mg活性化合物)。示例性的制剂10-片剂:可以将干粉形式的本发明化合物与干燥的凝胶粘合剂以约1:2的重量比混合。加入较少量的硬脂酸镁作为润滑剂。使该混合物在压片机中成型为270-450mg片剂(每个片剂含有90-150mg活性化合物)。使用和治疗方法如本文中一般性描述的,本发明涉及可充当例如GABA调节剂的神经活性类固醇。在某些实施方案中,预期这些化合物可用作治疗剂,用于治疗本文中所述的病症,例如,震颤(例如,特发性震颤);抑郁症(例如,产后抑郁),所述治疗包括对受试者施用有效量的本发明的化合物或其组合物。在某些实施方案中,通过静脉内施用来施用化合物。早期研究(参见,例如,Gee等人,EuropeanJournalofPharmacology,136:419-423(1987))证明,一些3α-羟基化的类固醇作为GABA受体复合物(GRC)的调节剂的效力,比已报道的其它物质高出更多数量级(参见,例如,Majewska等人,Science232:1004-1007(1986);Harrison等人,JPharmacol.Exp.Ther.241:346-353(1987))。Majewska等和Harrison等人认为,3α-羟基化的-5-还原的类固醇仅能够实现低得多的功效水平。体外和体内实验数据现已证明,这些类固醇的高效能容许它们在经由GRC的脑兴奋性的调节中是治疗上有用的(参见,例如,Gee等,EuropeanJournalofPharmacology,136:419-423(1987);Wieland等,Psychopharmacology118(l):65-71(1995))。多种合成类固醇还已被制备成神经活性类固醇。参见,例如,美国专利No.5,232,917,其公开了神经活性类固醇化合物,以治疗有益的方式用于治疗对GRC活性剂敏感(amenable)的以下病症:应激反应、焦虑症、失眠症、发作性疾病和心境障碍例如,抑郁症。此外,先前已证明,这些类固醇在GRC上的独特位点处相互作用,所述独特位点不同于其它已知的相互作用位点(例如,巴比妥酸盐、苯并二氮杂和GABA),在所述其它已知的相互作用位点中,先前已经得出对应激反应、焦虑症、睡眠、心境障碍和发作性疾病具有治疗有益效果(参见,例如,Gee,K.W.andYamamura,H.I.,“BenzodiazepinesandBarbiturates:DrugsfortheTreatmentofAnxiety,InsomniaandSeizureDisorders”,inCentralNervousSystemDisorders,Horvell,ed.,Marcel-Dekker,NewYork(1985),pp.123-147;Lloyd,K.G..andMorselli,P.L.,“PsychopharmacologyofGABAergicDrugs”,inPsychopharmacology:TheThirdGenerationofProgress,H.Y.Meltzer,ed.,RavenPress,N.Y.(1987),pp.183-195;和Gee等,EuropeanJournalofPharmacology,136:419-423(1987))。由于这些化合物的持续时间、效力和口服活性(以及其它施用形式),它们是合乎需要的。本文所描述的本发明化合物可以调节GABA功能,并因此在受试者中可以用作治疗和预防CNS相关病症的神经活性类固醇。本文使用的调节是指抑制或增强GABA受体功能。相应地,本发明提供的化合物和药物组合物在哺乳动物(包括人和非人哺乳动物)中用于治疗,预防和/或治疗CNS病症。由此,如前所述,本发明在其范围内包括并延伸至所列举的治疗方法、用于这种方法的化合物,以及该化合物用于制备这种方法所使用的药物的用途。与GABA调节相关的示例性的CNS病症包括但不限于:睡眠障碍[例如,失眠]、心境障碍[例如,抑郁症、精神抑郁病症(例如,轻度抑郁症)、双相性精神障碍(例如,I和/或II)、焦虑症(例如,广泛性焦虑症(GAD)、社会焦虑症)、应激反应、创伤后精神紧张性障碍(PTSD)、强制性障碍(例如,强迫性的强制性障碍(OCD))]、精神分裂症谱系障碍[例如,精神分裂症、情感分裂性精神障碍]、痉挛性障碍[例如,癫痫(例如,癫痫持续状态(SE))、癫痫发作]、记忆障碍和/或认知障碍[例如,注意力障碍(例如,注意缺陷多动障碍(ADHD))、痴呆(例如,阿尔茨海默型痴呆、Lewis体型痴呆、血管型痴呆]、运动障碍[例如,亨廷顿舞蹈症、帕金森氏症]、人格障碍[例如,反社会的人格障碍、强迫性的强制性人格障碍]、自闭症谱系障碍(ASD)[例如,自闭症、单病因的自闭症,例如突触机能障碍(synaptophathy),例如,Rett综合征、脆性X染色体综合征、Angelman综合征]、疼痛[例如,神经性疼痛、损伤相关的疼痛综合征、急性疼痛、长期疼痛]、外伤性脑损伤(TBI)、血管疾病[例如,中风、缺血、血管畸形]、物质滥用障碍和/或戒断综合征[例如,对阿片剂、可卡因和/或洒精成瘾]]和耳鸣。在又一个方面,提供了本发明的化合物与其它药理学活性剂的组合。本发明提供的化合物可以以单一活性剂形式施用,或它们可以与其它药剂一起组合施用。利用本领域技术人员显而易见的任何技术,包括,例如,单独、依次、同时和交替方式施用,可以进行组合施用。在另一方面,提供了治疗或预防对脑兴奋性有关的病症敏感或者被其折磨的受试者的脑兴奋性的方法,该方法包括:对受试者施用有效量的本发明的化合物。在另一方面,提供了治疗或预防受试者中的震颤的方法,其包括对需要此类治疗的受试者施用有效量的本发明化合物。在某些实施方案中,震颤是特发性震颤。在另一方面,提供了治疗或预防受试者中的心境障碍的方法,其包括对需要此类治疗的受试者施用有效量的本发明化合物。在某些实施方案中,心境障碍是抑郁症。在一些实施方案中,心境障碍是产后抑郁症。在另一方面,提供了缓解或预防受试者中PMS或PND的方法,其包括向需要此类治疗的受试者施用有效量的本发明化合物。在另一方面,提供了治疗或预防受试者的压力或焦虑的方法,其包括对需要此类治疗的受试者施用有效量的本发明化合物或其组合物。在又一个方面,提供了减轻或防止受试者失眠的方法,该方法包括:施用需要这种治疗的受试者有效量的本发明的化合物或其组合物。在又一方面,提供了诱发睡眠和基本上保持在正常睡眠中发现的REM睡眠水平的方法,其中未诱发实质的反弹性失眠,所述方法包括:施用有效量的本发明的化合物。在又一个方面,提供了通过对受试者施用治疗有效量的本发明的化合物来增强认知能力或治疗记忆障碍的方法。在一些实施方案中,所述障碍是阿尔茨海默病。在一些实施方案中,所述障碍是Rett综合征。在又一个方面,提供了通过对受试者施用治疗有效量的本发明的化合物来治疗注意力障碍的方法。在一些实施方案中,所述注意力障碍是ADHD。在一些实施方案中,长期对受试者施用所述化合物。在一些实施方案中,口服、皮下、肌肉内或静脉内对受试者施用所述化合物。焦虑症焦虑症是一个总称,包括若干不同形式的异常和病态恐惧以及焦虑。现行的精神病学诊断标准能够辨别多种焦虑症。广泛性焦虑症是常见的慢性病症,其特征为:焦虑状态持久,不能集中于任何一个目标或情况。患有广泛性焦虑症的患者感受到非特定性的持久性恐惧和烦恼,并且变得过度地关心日常事物。广泛性焦虑症是最常见的焦虑症,影响年长的成人。在恐慌病症中,遭受强烈恐怖和顾虑的暂时性发病的人,通常显现发抖、晃动、意识模糊、眩晕、恶心、呼吸困难。APA将这些恐慌发作定义为突然出现的恐惧或不适,在十分钟以内达到最高峰,可以持续几个小时,并且可以通过紧张、恐惧或甚至锻炼而引发;不过,具体病因并不总是很明显。除了复发性的意外恐慌发作之外,恐慌病症的诊断还要求所述发病具有长期结果:对发病的可能性烦恼、对将来的发病持久恐惧或与发病相关的行为出现显著变化。相应地,遭受恐慌病症的人感受到的症状甚至超出特定恐慌急性发作的范围。通常,恐慌患者注意到心跳的正常变化,使得他们认为他们的心脏有毛病,或他们即将出现另一次恐慌发作。在某些情况下,在恐慌发作期间,人体机能的意识增高(警觉过度),其中,任何可察觉的生理变化被认为是可能危及生命的疾病(即,极端臆想症)。强迫性的强制性障碍是焦虑症的一种类型,主要以重复性的强迫观念(痛苦、持久和侵入的想法或想象)和强迫症(迫切进行具体行动或习惯)为特征。可以在某种程度上把OCD思维模式比作迷信,它涉及一种相信事实上不存在的因果关系。通常,过程完全不合逻辑;例如,可能采用特定行走模式的强迫症,以减轻威胁伤害的强迫观念。在很多情况下,强迫症完全莫名其妙,简单地强迫完成神经所引发的习惯。在少数情况下,OCD患者可能只有强迫观念感受,没有明显的强迫症;更少数的患者只感受到强迫症。焦虑症的单一最大类型是恐惧症,它包括具体刺激或情况所引发的恐惧和焦虑的所有病例。患者从遇到他们恐惧的目标开始,典型地预测恐怖结果,所述目标可以是从动物到位置到体液的任何东西。创伤后精神紧张性障碍或PTSD是起因于外伤性经历的焦虑症。外伤后的应激反应可以起因于极端情况,例如,格斗、强奸、挟为人质或甚至严重事故。它还可以起因于长期接触严重的紧张性刺激,例如,能够忍受单一作战但不能应付连续作战的士兵。共同症状包括幻觉重现、逃避行为和压抑。进食障碍进食障碍特征在于饮食行为和体重调节的紊乱,并且与广泛的不良心理,身体和社会后果相关。患有进食障碍的个体可能开始只吃或少或多的食物,但在某个点时,他们失去控制或多或少渴望进食。进食障碍的特征可以是对体重或形状的严重苦恼或关注,或者极端努力来控制体重或食物摄入。进食障碍包括神经性厌食,神经性贪食,贪吃症,恶病质及其变型。患有神经性厌食的个体通常认为自己超重,即使在他们体重不足时。患有神经性厌食的个体可以对进食,食物和体重控制痴迷。患有神经性厌食的个体通常重复称重,仔细分配食物,并且仅食用非常少量的某些食物。患有神经性厌食的个体可以进行暴食,然后是极度节食,过度运动,自我诱导的呕吐或滥用泻药,利尿剂或灌肠剂。症状包括极低的体重,严重的食物限制,不断追求瘦弱和不愿意维持正常或健康的体重,强烈恐惧体重增加,扭曲身体形象和受体重和形状的感知严重影响的自尊,或否认低体重的严重性,女孩和妇女间缺乏月经。其它症状包括骨骼变薄,头发和指甲变脆,皮肤干燥和黄色,全身细毛生长,轻度贫血,肌肉萎缩和虚弱,严重便秘,低血压或呼吸缓慢和脉搏,损伤对心脏的结构和功能,脑损伤,多器官衰竭,体内温度下降,嗜睡,迟缓和不育。具有神经性贪食的个体具有食用异常大量的食物的重复和频繁事件,并且感觉缺乏对这些事件的控制。这种贪吃之后是补偿暴饮暴食的行为,例如强迫呕吐,过量使用泻药或利尿剂,禁食,过度运动或这些行为的组合。与神经性厌食不同,神经性贪食的人通常维持被认为是健康或正常体重的体重,而一些则略微超重。但是像神经性厌食的人一样,他们通常害怕增加体重,拼命想要减肥,并且对他们的身体大小和形状不满意。通常,贪食症行为是秘密做的,因为它往往伴随着厌恶或耻辱的感觉。贪吃和清洗周期可以发生在任何地方,从一周几次到一天多次。其它症状包括慢性发炎和咽喉痛,颈部和颌部肿胀的唾液腺,磨损的牙釉质和日益敏感和腐烂的牙齿(由于暴露于胃酸),酸反流障碍和其它胃肠道问题,肠道不适和来自轻泻剂滥用的刺激,来自清洗流体的严重脱水,电解质不平衡(其可导致心脏病发作或中风)。患有贪吃症的个体失去对其进食的控制。与神经性贪食不同,贪吃的时期之后不会进行代偿性行为,如清洗,过度运动或禁食。患有贪吃症的个体通常是超重或肥胖。患有贪吃症的肥胖个体具有发展心血管疾病和高血压的较高风险。他们也经历过关于他们的贪吃的内疚,羞耻和痛苦,这可以导致更多的贪吃。恶病质也称为“消耗性病症(wastingdisorder)”,并且是许多癌症患者经历的与饮食有关的问题。患有恶病质的个体可以继续正常地进食,但是他们的身体可能拒绝利用它摄取的维生素和营养物,或者他们将失去食欲并停止进食。当个体经历食欲减退和停止进食时,可以认为他们已经产生神经性厌食。神经变性疾病和障碍术语“神经变性疾病”包括与神经元的渐进性的结构或功能的丧失或神经元的死亡有关的疾病和障碍。神经变性疾病和障碍包括但不限于:阿尔茨海默病(包括轻度、中度或重度的认知损害的相关症状);肌萎缩性侧索硬化(ALS);缺氧性的和缺血性的损伤;运动失调和惊厥(包括:情感分裂性精神障碍或用于治疗精神分裂症谱系障碍的药物所引起的癫痫发作的治疗和预防所造成的);良性健忘;脑水肿;小脑性共济失调,包括McLeod神经棘红细胞增多综合征(MLS);封闭性的颅脑损伤;昏迷;挫伤性损伤(例如,脊髓损伤和颅脑损伤);痴呆,包括多梗塞性痴呆和老年痴呆;意识障碍;唐氏综合征;药物诱导或药物治疗诱导的震颤性麻痹(例如,抗精神病药诱导的急性静坐不能、急性肌张力障碍、震颤性麻痹或迟发性运动障碍、神经阻滞剂恶性综合征或药物诱导的姿势性震颤);癫痫;脆性X染色体综合征;GillesdelaTourette综合征;头部创伤;听力损伤和丧失;亨廷顿舞蹈症;Lennox综合征;左旋多巴诱导的运动障碍;智力迟钝;运动障碍,包括运动不能症和运动不能(僵硬)综合征(包括基底神经节钙化、皮质基底核退化、多系统萎缩、震颤性麻痹-ALS痴呆复合体、帕金森氏症、脑炎后震颤麻痹和渐进性核上性麻痹);与肌原纤维僵直或无力相关的肌肉痉挛和病症,包括舞蹈病(例如,良性遗传性的舞蹈病、药物诱导的舞蹈病、偏侧颤搐、亨廷顿舞蹈症、神经棘红细胞增多症、Sydenham舞蹈病和症状性舞蹈病)、运动障碍(包括抽搐,例如,复杂型抽搐、简单抽搐和症状性抽搐)、肌阵挛(包括全身性肌阵挛和局灶性肌阵挛)、震颤(例如,休息性震颤、姿势性震颤和意向震颤)和肌张力障碍(包括轴性肌张力障碍、张力障碍性指痉挛、偏瘫性肌张力障碍、突发性的肌张力障碍和局灶性肌张力障碍,例如睑痉挛、口下颌肌张力障碍和间歇性的言语障碍和斜颈);神经元损伤,包括眼睛的视觉损伤、视网膜病或黄斑变性;大脑中风、血栓栓塞性中风、出血性中风、脑缺血、脑血管痉挛、血糖过低、健忘症、低氧症、缺氧症、出生时的窒息和心跳停止之后的神经损伤;帕金森氏症;癫痫发作;癫痫持续状态;中风;耳鸣;小管硬化症和病毒感染诱导的神经变性(例如,爱滋病(AIDS)和脑病所引起的神经变性)。神经变性疾病还包括但不限于:大脑中风、血栓栓塞中风、出血性中风、脑缺血、脑血管痉挛、血糖过低、健忘症、低氧症、缺氧症、出生时的窒息和心跳停止之后的神经损伤。治疗或预防神经变性疾病的方法还包括:治疗或预防神经变性病症特有的神经元功能的丧失。癫痫癫痫是以随着时间的推移而重复的癫痫发作为特征的脑病。癫痫的类型可以包括但不限于:普遍性癫痫,例如,儿童癫痫小发作、少年肌肉阵挛性癫痫、觉醒时的癫痫大发作、West综合征、肌阵挛性起立不能小发作、部分性癫痫,例如,精神运动性癫痫、额叶癫痫、儿童期的良性局灶性癫痫。癫痫持续状态(SE)癫痫持续状态(SE)可以包括,例如,惊厥性癫痫持续状态,例如,早期癫痫持续状态、完全的癫痫持续状态、难治疗的癫痫持续状态、超难治疗的癫痫持续状态;非抽搐性癫痫持续状态,例如,全身性持续状态、复杂的部分癫痫持续状态;全身性周期性癫痫样放电;以及周期性一侧(性)癫痫样放电。惊厥性癫痫持续状态的特征在于:存在惊阙性癫痫持续状态的癫痫发作,并且可以包括早期癫痫持续状态、确定的癫痫持续状态、难治疗的癫痫持续状态、超难治疗的癫痫持续状态。可用一线疗法来治疗早期癫痫持续状态。完全的癫痫持续状态的特征在于:癫痫持续状态发作,虽然用一线疗法和二线疗法治疗,但还持续发作。难治疗的癫痫持续状态的特征在于:癫痫持续状态发作,虽然用一线疗法和二线疗法治疗,但还持续发作,并且通常进行全身麻醉。超难治疗的癫痫持续状态的特征在于:癫痫持续状态发作,虽然用一线疗法、二线疗法和全身麻醉进行治疗,但还持续发作24小时或更长时间。非惊厥性癫痫持续状态可以包括,例如,局灶性非惊厥性癫痫持续状态,例如,复杂的部分非惊厥性癫痫持续状态、简单的部分非惊厥性癫痫持续状态、轻度非惊厥性癫痫持续状态、全身性非惊厥性癫痫持续状态,例如,迟发性失神性非惊厥性癫痫持续状态、非典型失神性非惊厥性癫痫持续状态或典型失神性非惊厥性癫痫持续状态。还可以在发作之前,对患有下列病症的受试者施用本文所描述的组合物,以作为预防剂:CNS病症,例如,外伤性脑损伤、癫痫持续状态,例如,惊厥性癫痫持续状态,例如,早期癫痫持续状态、完全的癫痫持续状态、难治疗的癫痫持续状态、超难治疗的癫痫持续状态;非惊厥性癫痫持续状态,例如,全身性持续状态、复杂的部分癫痫持续状态;全身性周期性癫痫样放电;以及周期性一侧(性)癫痫样放电。发作(Seizure)发作是在脑中的异常电活性的发作之后出现的物理发现或行为变化。术语“发作”通常可与“惊厥”互换使用。当人的身体快速并且不能控制地晃动时,是惊厥。在惊厥期间,人的肌肉反复地收缩和松驰。基于行为和大脑活动的类型,发作被分成两个大类:全身性和部分性(还称为局部或局灶性)发作。发作类型的分类,能够帮助医生诊断患者是否患有癫痫。电脉冲遍及整个脑部,引起全身性发作,而电脉冲在脑的相对小部分中,则引起部分性发作(至少在初始的时候)。产生发作的脑的部分有时称为病灶。全身性发作有六个类型。最常见的和突出的,并因此是最众所周知的,是全身性惊厥,还称为大发作。在这类发作中,患者丧失意识,并且通常崩溃。意识丧失之后,持续全身性身体变硬(称为发作的“强直”阶段)30至60秒,然后强烈痉挛(“阵挛”阶段)30至60秒,而后患者深度睡眠(“发作后”或发作之后的阶段)。在大发作期间,可能出现损伤和意外,例如,咬舌和尿失禁。失神性发作导致短时意识丧失(只有几秒),同时几乎没有症状。患者(通常大部分是儿童)典型地中止活动,并且发呆。这些发作突然地开始和结束,并且一天可以出现若干次。患者通常没有意识到他们出现发作,只不过他们可以意识到“失去时间”。肌肉阵挛性发作包括偶发性痉挛,通常在身体的两侧。患者有时将痉挛描述为暂时性电震。当发作强烈时,这些发作可以导致跌倒,或无意地投掷物体。阵挛性发作是重复的、有节奏的痉挛,它同时涉及身体的两侧。强直性发作的特征在于肌肉变硬。无张力性发作包括肌张力的突然和全身性丧失,尤其是臂和腿,通常导致跌倒。本文描述的发作可以包括:癫痫性发作;急性的重复发作;集群性发作;连续发作;不间断的发作;长期发作;复发性发作;癫痫持续状态,例如,难治疗的惊厥性癫痫持续状态、非惊厥性癫痫持续状态;难治疗的发作;肌肉阵挛性发作;强直性发作;强直-阵挛性发作;简单的部分性发作;复杂的部分性发作;继发性全身性发作;非典型性的失神性发作;失神性发作;无张力性发作;良性Rolandic发作;热性发作;情感性发作;局灶性发作;痴笑性发作;全身性发作;婴幼儿痉挛;病灶性发作;大规模的双向肌阵挛性发作;多灶性发作;初生儿发作;夜间发作;枕叶发作;外伤后的发作;微小发作;Sylvan发作;视反射性发作;或撤药痉挛。震颤震颤是不自主的(有时是节奏性的)肌肉收缩和放松,其可以涉及一个或多个身体部位(例如,手,手臂,眼,脸,头,声带,躯干,腿)的振动或颤搐。小脑震颤或意向性震颤是在有目的的运动之后发生的肢体的缓慢,广泛的震颤。小脑震颤是由例如肿瘤,中风,疾病(例如,多发性硬化,遗传性变性性病症)引起的小脑中的损伤或对小脑的损伤引起的。肌张力不全震颤(dystonictremor)发生在受张力失常(一种运动障碍,其中持续的无意识肌肉收缩导致扭转和重复运动和/或疼痛和异常的姿势或位置的)影响的个体中。肌张力不全震颤可能影响身体内的任何肌肉。肌张力不全震颤不规则地发生,并且经常可以通过完全休息来缓解。特发性震颤或良性特发性震颤是最常见的震颤类型。特发性震颤可以是轻度的,并且一些是非进展性的,并且可以是缓慢进展的,从身体的一侧开始,但在3年内影响两侧。手最常受影响,但头部,声音,舌,腿和躯干也可能累及。震颤频率可能随着人的年龄下降,但严重程度可能会增加。加重的情绪,压力,发烧,身体疲惫或低血糖可引发震颤和/或增加其严重性。站立性颤抖(orthostatictremor)的特征在于快速(例如,大于12Hz)节律性肌肉收缩,其在站立后立即在腿和躯干中发生。在大腿和腿部感觉到痉挛,并且当被要求站在一个地点时,患者可能无法控制地摇晃。站立性颤抖可发生在患有特发性震颤的患者中。帕金森病震颤(parkinsoniantremor)是由对大脑内控制运动的结构的损伤引起的。帕金森病震颤通常是帕金森病的前兆,并且通常被视为手的“滚丸”动作,也可能影响下巴,唇,腿和躯干。帕金森病震颤的发作通常在60岁后开始。运动在一个肢体或身体的一侧开始,并且可以进展到包括另一侧。生理性震颤可发生在正常个体中并且没有临床意义。它可以在所有自愿的肌肉群中看到。生理性震颤可由某些药物,酒精戒断或包括活动过度的甲状腺和低血糖的医学状况引起。震颤通常具有约10Hz的频率。心理性震颤或歇斯底里震颤可发生在休息或姿势或动力运动期间。患有心理性震颤的患者可能具有转变障碍或另一种精神疾病。红核性震颤(rubraltremor)的特征在于粗略的慢震颤,其可以在静息,姿势和在意图的情况下存在。震颤与影响中脑中的红核的状况,经典的不寻常中风相关。心境障碍临床抑郁症也称为重性抑郁症,重性抑郁障碍(MDD),重度抑郁症,单相抑郁症,单相障碍和复发性抑郁症,并且是指以普遍持续的低情绪为特征的精神障碍,伴有低自尊心和在正常的愉快的活动中的兴趣或快乐的丧失。有些临床抑郁症患者睡眠困难,体重减轻,一般感到焦虑和烦躁。临床抑郁症影响个人的感觉,思考和行为,并可能导致各种情绪和身体问题。患有临床抑郁症的个体可能难以进行日常活动,并使个体感觉如同生命不值得活一样。产后抑郁(PND)也称为产后抑郁症(PPD),并且是指一种在分娩后影响女性的临床抑郁症。症状可以包括悲伤,疲劳,睡眠和饮食习惯的改变,性欲减退,哭泣发作,焦虑和易怒。在一些实施方案中,PND是治疗抗性抑郁症(例如,如本文所述的治疗抗性抑郁症)。在一些实施方案中,PND是难治性抑郁症(例如,如本文所述的难治性抑郁症)。非典型抑郁(AD)的特征在于情绪反应性(例如,矛盾的兴趣缺失(paradoxicalanhedonia))和积极性,显著的体重增加或食欲增加。患有AD的患者还可能具有过度的睡眠或嗜睡(睡眠过度),肢体重度的感觉和作为对感知的人际间排斥的超敏性的结果的显著的社会障碍。忧郁性抑郁症(Melancholicdepression)的特征在于在大多数或所有活动中丧失快乐(兴趣缺失),对愉悦刺激反应失败,抑郁情绪比悲伤或损失情绪更明显,过度体重减轻或过度内疚。精神性重性抑郁症(PMD)或精神病性抑郁症是指重性抑郁发作,特别是忧郁症性质,其中个体经历精神病症状,例如妄想和幻觉。紧张性抑郁症是指涉及运动行为和其它症状紊乱的重性抑郁症。个体可能变得沉默和木僵,并且是不动的或展示无意义或奇怪的运动。季节性情感障碍(SAD)是指一种季节性抑郁症类型,其中个体在秋季或冬季有抑郁发作的季节性模式。心境恶劣是指与单相抑郁相关的状况,其中相同的身体和认知问题是明显的。它们不那么严重,往往更持久(例如,至少2年)。双重抑郁(Doubledepression)指持续至少2年的相当抑郁的情绪(恶劣心境),并且由重度抑郁症的时期分隔。抑郁性人格障碍(DPD)是指具有抑郁特征的人格障碍。复发性短暂性抑郁(RBD)是指个体具有约每月一次的抑郁发作的状况,每次发作持续2周或更少,通常小于2-3天。轻度抑郁症或轻微抑郁是指其中存在至少2种症状2周的抑郁症。双相障碍或躁郁症(manicdepressivedisorder)引起极度情绪波动,包括情绪高(躁狂或轻躁狂)和低(抑郁)。在躁狂期间,个体可能感觉或行为异常愉快,精力充沛或易怒。他们经常做出很少考虑的决定,很少考虑到结果。睡眠的需要通常减少。在抑郁症期间,可能会哭泣,与他人很少眼睛接触,以及对生活的负面观点。20年来,患有该疾病的人中自杀的风险较高,达6%以上,而自身伤害发生在30-40%中。其它精神健康问题如焦虑症和物质使用障碍通常与双相型障碍有关。由慢性医学状况引起的抑郁症是指由慢性医学状况如癌症或慢性疼痛,化疗,慢性压力引起的抑郁症。治疗抗性抑郁症是指已经对个体治疗抑郁症但是症状没有改善的状况。例如,抗抑郁药或心理咨询(心理治疗)不能缓解患有治疗抗性抑郁症的个体的抑郁症状。在一些情况下,具有治疗抗性抑郁症的个体的症状改善,但复发。难治性抑郁症发生在对标准药理治疗包括三环抗抑郁药,MAOI,SSRI,以及双重和三重摄取抑制剂和/或抗焦虑药,以及非药理治疗(例如,心理治疗,电惊厥治疗,迷走神经刺激和/或经颅磁刺激)有抗性的患有抑郁症的患者中。自杀性,自杀意念,自杀行为是指个人自杀的倾向。自杀意念涉及对自杀的想法或不寻常的关注。自杀意念的范围极大变化,从例如短暂的想法到广泛的想法,详细的计划,角色扮演,不完全的尝试。症状包括谈论自杀,获得自杀的手段,退出社交联系,关注死亡,感觉陷入困境或无望的情况,增加使用酒精或毒品,做风险或自我毁灭的事情,告别别人就像他们不会再见面一样。抑郁的症状包括持续的焦虑或悲伤的感觉,无助的感觉,无望,悲观主义,无价值,低能量,不安,易怒,疲劳,对兴趣活动或爱好的兴趣丧失,缺乏积极的想法或计划,过度睡眠,进食过量,食欲下降,失眠,自我伤害,自杀想法和自杀企图。症状的存在,严重性,频率和持续时间可以根据具体情况而变化。抑郁症的症状和其缓解可由内科医生或心理学家(例如,通过精神状态检查)确定。麻醉/镇静麻醉是药理学诱导的和可逆性的健忘、痛觉丧失、响应缺失、骨骼肌反射缺失、应激反应降低或所有这些同时存在的状态。这些效果可以从能够单独提供合适的组合效果的单一药物获得,或偶而使用组合药物(例如,安眠药、镇静剂、麻痹剂、镇痛药),获得非常特定的组合效果。麻醉允许患者进行手术及其它过程,使他们不会另外经受痛苦和疼痛。镇静是通过施用药理学药剂而使应激性或兴奋性降低,通常便于医学过程或诊断过程。镇静和镇痛包括连续的神智清醒的状态,范围从轻度镇静(焦虑症)至全身麻醉。轻度镇静亦称抗焦虑。轻度镇静是药物诱导的状态,在此期间,患者通常对语言命令有反应。认知功能和协调性可能受到削弱。换气和心血管功能典型地未受影响。中度镇静/镇痛(清醒性镇静)是药物诱导的意识抑制状态,在此期间,患者对单独或伴有轻微触觉刺激的语言命令有目的地反应。通常不需要干预来保持呼吸道开放。自发性换气典型地可以满足要求。通常保持心血管功能。深度镇静/镇痛是药物诱导的意识抑制状态,在此期间,不容易唤醒患者,但在重复或疼痛刺激之后,会有目的地反应(疼痛刺激没有产生反射性退缩)。独立的通气功能可能受到削弱,并且为了保持呼吸道开放,患者可能需要得到帮助。自发性换气可能不足。通常保持心血管功能。全身麻醉是药物诱导的意识丧失,在此期间,不能唤醒患者(甚至用疼痛刺激)。保持独立通气功能的能力通常受到削弱,并且为了保持呼吸道开放,通常需要帮助。由于自发性换气受到抑制,或药物诱导的神经肌肉机能降低,所以,可能需要正压通气。心血管功能可能受到削弱。在重病监护室(ICU)中,镇静状态使得患者对环境的意识降低,并且降低他们对外部刺激的反应。它可以在护理病危患者中起作用,并且包括宽范围的症状控制,所述症状将在不同患者之间以及在个体中在其患病期间发生变化。在病危护理中,使用深度镇静状态,便于气管内导管耐受性和通气同步性,通常使用神经肌肉阻断剂。在一些实施方案中,在ICU中,诱导镇静状态(例如,长期镇静状态、持续镇静状态),并长时间保持(例如,1天、2天、3天、5天、1周、2周、3周、1个月、2个月)。长效镇静药剂可以作用长时间。在ICU中,镇静药剂可以具有短的消除半衰期。程序性镇静和镇痛还称为清醒性镇静,是施用镇静剂或分离麻醉剂且含有或不含有镇痛药的技术,诱导受试者忍受不愉快的过程的状态,同时保持心肺功能。实施例为了可以更充分地理解本文描述的发明,列出下列实施例。提供本申请所描述的合成和生物学实施例,用来说明本发明提供的化合物、药物组合物和方法,且其不以任何方式解释为限制它们的范围。原料和方法使用下列一般方法和工艺,由容易获得的起始原料可以制备本发明提供的化合物。应理解,在施用的典型的或优选的工艺条件下(即,反应温度、时间、反应物的摩尔比、溶剂、压力,等),还可以使用其它工艺条件,除非另有说明。最佳反应条件可以随所使用的具体反应物或溶剂而变化,但这种条件可以由本领域技术人员通过常规优化过程来确定。另外,对本领域技术人员显而易见的是,可能需要常规保护基,以防止某些官能团进行不希望的反应。对于具体官能团的合适保护基团以及对于保护和脱保护的合适条件的选择在本领域中是公知的。例如,许多保护基团以及它们的引入和除去描述在T.W.GreeneandP.G.M.Wuts,ProtectingGroupsinOrganicSynthesis,SecondEdition,Wiley,NewYork,1991及其中引用的参考文献中。本发明提供的化合物可通过已知的标准方法分离和纯化。这种方法包括(但不限于)重结晶、柱色谱法、HPLC或超临界流体色谱法(SFC)。关于本文列出的代表性的取代的三唑或四唑的制备细节,提供下列反应路线。本发明提供的化合物,可通过有机合成领域的技术人员由已知的或商购的原料和试剂制备。在本发明提供的对映异构体/非对映异构体的分离/纯化过程中,可以使用的示例性的手性柱包括但不限于:AD-10、OB、OB-H、OD、OD-H、OF、OG、OJ和OK。本文报道的1H-NMR(例如对于中间体)可以是化合物(例如,本文所描述的化合物)的全部NMR谱的部分表示。例如,报道的1HNMR可以不包括大约1至大约2.5ppm的δ(ppm)之间的区域。代表性实施例的全部1H-NMR谱的备份提供于附图中。制备型HPLC的示例性一般方法:柱:WatersRBridge制备型10μmC18,19*250mm。移动相:乙腈、水(NH4HCO3)(30L水,24gNH4HCO3,30mLNH3.H2O)。流速:25mL/min分析型HPLC的示例性一般方法:移动相:A:水(10mMNH4HCO3),B:乙腈。梯度:5%-95%B,1.6或2分钟。流速:1.8或2mL/min;柱:XBridgeC18,4.6*50mm,3.5μm,45℃。合成方法本发明的化合物可按照本领域描述的方法(Upasani等,J.Med.Chem.1997,40:73-84;和Hogenkamp等,J.Med.Chem.1997,40:61-72),并使用合适的试剂、原料和本领域技术人员已知的纯化方法制备。在一些实施方案中,可以使用常规反应路线1-3中所示的方法,其包括用亲核试剂对19-去甲孕烷溴化物进行亲核取代,制备本文所描述的化合物。在一些实施方案中,在K2CO3的存在下,在THF中,亲核试剂与19-去甲孕烷溴化物反应。反应路线1反应路线2反应路线3实施例1:合成SA和SA中间体合成化合物SA-B。将化合物SA-A(50克,184mmol)和钯黑(2.5克)(在四氢呋喃(300mL)中)和浓氢溴酸(1.0mL)用10atm氢气氢化。在室温搅拌24小时之后,通过硅藻土垫过滤该混合物,并将滤液真空浓缩,提供粗品化合物。用丙酮重结晶,得到化合物SA-B(42.0克,产率:83.4%)的白色粉末。1HNMR:(400MHz,CDCl3)δ2.45-2.41(m,1H),2.11-3.44(m,2H),3.24(s,3H),2.18-2.15(m,1H),2.01-1.95(m,1H),1.81-1.57(m,7H),1.53-1.37(m,7H),1.29-1.13(m,3H),1.13-0.90(m,2H),0.89(s,3H)。合成化合物SA-C。在氮气氛围中,在-78℃,将SA-B(42.0克,153.06mmol)的600mL无水甲苯溶液逐滴加入到甲基铝二(2,6-二叔丁基-4-甲基酚盐(methylaluminumbis(2,6-di-tert-butyl-4-methylphenoxide)(MAD)(459.19mmol,3.0eq,新制备)溶液中。加入完成之后,将该反应混合物在-78℃搅拌1小时。然后,在氮气氛围中,在-78℃,将3.0MMeMgBr(153.06mL,459.19mmol)慢慢地逐滴加入到上述混合物中。然后,将该反应混合物在此温度搅拌3小时。TLC(石油醚/乙酸乙酯=3:1)显示反应完成。然后,在-78℃,将饱和NH4Cl水溶液慢慢地逐滴加入到上述混合物中。加入完成之后,过滤该混合物,用EtOAc洗涤滤饼,用水和盐水洗涤有机层,用无水Na2SO4干燥,过滤,浓缩,用硅胶快速色谱纯化(石油醚/乙酸乙酯,20:1至3:1),提供化合物SA-C(40.2克,产率:90.4%)的白色粉末。1HNMR:(400MHz,CDCl3)δ2.47-2.41(m,1H),2.13-2.03(m,1H),1.96-1.74(m,6H),1.70-1.62(m,1H),1.54-1.47(m,3H),1.45-1.37(m,4H),1.35-1.23(m,8H),1.22-1.10(m,2H),1.10-1.01(m,1H),0.87(s,3H)。合成化合物SA-D。在0℃,向PPh3EtBr(204.52g,550.89mmol)的THF(500mL)溶液中加入t-BuOK(61.82g,550.89mmol)的THF(300mL)溶液。加入完成之后,将该反应混合物在60℃搅拌1小时,然后在60℃,逐滴加入溶于THF(300mL)中的SA-C(40.0g,137.72mmol)。将该反应混合物加热至60℃,保持18小时。将该反应混合物冷却至室温,用饱和NH4Cl淬灭,并用EtOAc(3*500mL)提取。将组合的有机层用卤水洗涤,干燥,浓缩,得到粗品,将其用快速柱色谱纯化(石油醚/乙酸乙酯,50:1至10:1),提供化合物SA-D(38.4g,产率:92%)的白色粉末。1HNMR:(400MHz,CDCl3)δ5.17-5.06(m,1H),2.42-2.30(m,1H),2.27-2.13(m,2H),1.89-1.80(m,3H),1.76-1.61(m,6H),1.55-1.43(m,4H),1.42-1.34(m,3H),1.33-1.26(m,6H),1.22-1.05(m,5H),0.87(s,3H)。合成化合物SA-E。在冰浴中,向SA-D(38.0g,125.62mmol)的无水THF(800mL)溶液中逐滴加入BH3.Me2S溶液(126mL,1.26mol)。加入完成之后,将该反应混合物在室温(14-20℃)搅拌3小时。TLC(石油醚/乙酸乙酯,3:1)显示反应完成。将该混合物冷却至0℃,并加入3.0M的NaOH水溶液(400mL),而后加入30%的H2O2水溶液(30%,300mL)。将该混合物在室温(14-20℃)搅拌2小时,而后过滤,并用EtOAc(3*500mL)提取。将组合的有机层用饱和Na2S2O3水溶液、卤水洗涤,用Na2SO4干燥,真空浓缩,得到粗品(43g,粗品)的无色油。粗品不用进一步纯化,在下一步中使用。合成化合物SA-F。在0℃,向SA-E(43.0g,134.16mmol)的二氯甲烷(800mL)溶液中分为几份加入PCC(53.8g,268.32mmol)。然后,将该反应混合物在室温(16-22℃)搅拌3小时。TLC(石油醚/乙酸乙酯,3:1)显示反应完成,然后过滤该反应混合物,并用DCM洗涤。用饱和Na2S2O3水溶液、卤水洗涤有机相,用Na2SO4干燥,并真空浓缩,得到粗品。用快速柱色谱纯化粗品(石油醚/乙酸乙酯,50:1至8:1),提供化合物SA-F(25.0g,产率:62.5%,两步)的白色粉末。1HNMR(SA-F):(400MHz,CDCl3)δ2.57-2.50(m,1H),2.19-2.11(m,4H),2.03-1.97(m,1H),1.89-1.80(m,3H),1.76-1.58(m,5H),1.47-1.42(m,3H),1.35-1.19(m,10H),1.13-1.04(m,3H),0.88-0.84(m,1H),0.61(s,3H)。合成化合物SA。向SA-F(10g,31.4mmol)和HBr水溶液(5滴,48%,在水中)的200mLMeOH溶液中逐滴加入溴(5.52g,34.54mmol)。将该反应混合物在17℃搅拌1.5小时。在0℃,将得到的溶液用饱和NaHCO3水溶液淬灭,并用EtOAc(150mLx2)提取。将组合的有机层干燥,并浓缩。用硅胶柱色谱纯化残余物,用(PE:EA=15:1至6:1)洗脱,提供化合物SA(9.5g,产率:76.14%)的灰白色固体。LC/MS:rt5.4min;m/z379.0,381.1,396.1。实施例2:合成化合物SA-1。向THF(5mL)中的K2CO3(25mg,0.18mmol)的悬浮液添加3H-1,2,4-三唑(32mg,0.46mmol)和SA(36mg,0.09mmol)。在rt搅拌混合物24小时。将反应混合物倒入5mLH2O中,并且用EtOAc(2×10mL)提取。用卤水(brine)清洗组合的有机层,通过硫酸钠干燥,过滤并且浓缩。通过反相prep-HPLC纯化残留物以提供标题化合物,为灰白色固体(11mg,31.3%)。1HNMR(400MHz,CDCl3),δ(ppm),7.67(s,1H),7.64(s,1H),5.27(AB,1H),4.18(AB,1H)2.65(1H,t),1.27(s,CH3),0.67(s,3H)。实施例3:合成化合物SA-2。向THF(5mL)中的K2CO3(25mg,0.18mmol)悬浮液添加1H-四唑(16mg,0.23mmol)和SA(70mg,0.09mmol)。在rt将混合物搅拌15h。将反应混合物倒入5mLH2O,并且用EtOAc(2×10mL)提取。用卤水清洗组合的有机层,通过硫酸钠干燥,过滤并且浓缩。通过反相prep-HPLC纯化残留物以提供标题化合物,为灰白色固体,SA-2(8mg,11.7%),和副产物(10mg,14.0%)。SA-2:1HNMR(500MHz,CDCl3),δ(ppm),8.74(s,1H),5.31(AB,1H),5.17(AB,1H),2.65(1H,t),1.28(s,CH3),0.67(s,3H)。实施例4:合成化合物SA-3。向DMF(20mL)中的SA(1g,2.52mmol)的悬浮液添加K2CO3(1.04g,7.55mmol)和4-甲基-2H-1,2,3-三唑(313.64mg,3.77mmol)。在室温将混合物搅拌3h。然后,将反应混合物倒入5mLH2O,并且用EtOAc(30mL)提取。用卤水清洗组合的有机层(10mL*3),通过硫酸钠干燥,过滤并且浓缩。通过prep-HPLC纯化残留物以提供标题化合物SA-3(269.2mg,收率=26.59%),为灰白色固体。1HNMR(SA-3)(400MHz,CDCl3)δ7.42(s,1H),5.14-5.13(m,2H),2.57-2.56(m,1H),2.33(s,3H),2.01-2.00(m,2H),1.81-1.70(m,6H),1.45-1.39(m,7H),1.27-1.24(m,9H),1.01-1.00(m,3H),0.70(s,3H)。实施例5:合成化合物SA-4和SA-5。在室温(13-17℃)在N2下向DMF(20mL)中的4-甲基-2H-1,2,3-三唑(836.4mg,10.07mmol)和K2CO3(1.39g,10.07mmol)溶液添加化合物SA(2.0g,5.03mmol)。在室温(13-17℃)将反应混合物搅拌4h。TLC显示完成了反应。然后,将反应混合物倒入水中,并且用EtOAc(50mLx3)提取。用卤水清洗组合的有机层,通过无水Na2SO4干燥,过滤并且在真空中浓缩。通过硅胶纯化残留物以提供SA-4/SA-5和副产物(500mg,收率:25%)的730mg混合物。通过SFC纯化分开混合物以给出SA-4(249.8mg,收率:12.5%)和SA-5(426.2mg,收率:21.3%),为灰白色固体。1HNMR(SA-4):(400MHz,CDCl3)δ7.49(s,1H),5.14-5.02(m,2H),2.67-2.63(m,1H),2.21-2.16(m,4H),2.11-2.08(m,1H),1.88-1.75(m,6H),1.65-1.55(m,1H),1.51-1.37(m,7H),1.33-1.22(m,8H),1.14-1.08(m,3H),0.69(s,3H).1HNMR(SA-5):(400MHz,CDCl3)δ7.35(s,1H),5.20-5.04(m,2H),2.65-2.61(m,1H),2.38(s,3H),2.25-2.17(m,1H),2.09-2.05(m,1H),1.88-1.63(m,7H),1.50-1.28(m,15H),1.15-1.06(m,3H),0.67(s,3H)。实施例6:合成化合物SA-6和SA-7。向THF(3mL)中的化合物SA(120mg,0.29mmol)溶液添加K2CO3(210mg,1.5mmol)和5-甲基-2H-四唑(126mg,1.5mmol)。在室温将所得的溶液搅拌过夜,此时LCMS分析显示反应完成。然后,用EtOAc(20mL)稀释反应,并且用卤水(10mL)清洗所得的溶液,通过Na2SO4干燥并且在真空中浓缩。通过prep-HPLC纯化残留物以给出SA-6(10mg,0.025mmol,收率=8%),SA-7(8mg,0.020mmol,收率=7%),为灰白色固体。SA-6:1HNMR(400MHz,CDCl3)δ5.16-5.03(m,2H),2.66(t,1H),2.46(s,3H),2.25-2.10(m,1H),2.08-2.02(m,1H),1.90-1.70(m,7H),1.68-1.02(m,18H),0.67(s,3H).LC-MS:rt=2.20min;m/z=401.3(M+H)+.SA-7:1HNMR:(400MHz,CDCl3)δ5.40-5.30(m,2H),2.62(t,1H),2.55(s,3H),2.30-2.00(m,2H),1.90-1.56(m,7H),1.50-1.02(m,18H),0.70(s,3H).LC-MS:rt=2.30min;m/z=401.2(M+H)+实施例7:合成化合物SA-8。在N2下在室温(14-20℃).向干DMF(10mL)中的化合物SA(150mg,0.377mmol)和K2CO3(104.3mg,0.755mmol)的溶液添加5-(三氟甲基)-1H-四唑(104.2mg,0.755mmol)。在相同的温度将反应混合物搅拌18h。将反应混合物倒入水,用EtOAc(50mLx3)提取。用卤水清洗有机层,通过无水Na2SO4干燥,过滤并且在真空中浓缩。通过硅胶柱纯化残留物(PE:EtOAc=10:1至1:1)以提供SA-8(89.1mg,收率:51.9%),为白色粉末。1HNMR(SA-8):(400MHz,CDCl3)δ5.51(s,2H),2.69-2.65(m,1H),2.26-2.18(m,1H),2.09-2.05(m,1H),1.87-1.77(m,6H),1.69-1.62(m,1H),1.55-1.43(m,7H),1.37-1.26(m,8H),1.19-1.09(m,3H),0.72(s,3H)。实施例8:合成化合物SA-9。向THF(5mL)中的K2CO3(25mg,0.18mmol)悬浮液添加3H-1,2,4-三唑(16mg,0.23mmol)和SA(70mg,0.09mmol)。在rt将混合物搅拌15h。将反应混合物倒入5mLH2O中,并且用EtOAc(2×10mL)提取。用卤水清洗组合的有机层,通过硫酸钠干燥,过滤并且浓缩。通过反相prep-HPLC纯化残留物以提供标题化合物,为灰白色固体,SA-9:1HNMR(400MHz,CDCl3),δ(ppm),7.76(s,1H),7.64(s,1H),5.27(AB,1H),5.14(AB,1H),2.65(1H,t),1.27(s,3H),0.67(s,3H)。实施例9:合成SC-SS和SC-SS中间体合成化合物SC-KK和SC-LL。向200mLDMSO中的三甲基碘化亚砜(trimethylsufoxoniumiodide)(43g,210mmol)的搅拌溶液添加NaH(60%,8.4g,210mmol)。在室温搅拌1h后,逐滴添加20mLDMSO中的化合物SC(30g,105mmol)的悬浮液。在2.5h后,将反应混合物倒入冰冷的水中,并且用乙酸乙酯(100mLx3)提取。然后,用卤水(100mLx3)清洗组合的乙酸乙酯层,用MgSO4干燥,过滤,并且浓缩。通过柱层析纯化残留物(石油醚/乙酸乙酯=20:1至15:1)以提供化合物SC-LL(14.7g,49mmol,47%)。合成化合物SC-MM和SC-NN。对反应物混合物SA-KK和SA-LL的混合物(3.0g,10.0mmol,1:1)添加干(Bu)4NF,然后,将混合物加热100℃过夜。将残留混合物倒入50mLH2O中,并且用EtOAc(2×50mL)提取。用卤水清洗组合的有机层溶液,通过硫酸钠干燥,过滤并且浓缩。通过快速层析(flashchromatography)纯化残留物(石油醚/乙酸乙酯=20:1)以提供产物混合物SC-MM和SC-NN(2.1g,6.5mmol,65%),为灰白色固体。合成化合物SC-OO和SC-PP。向无水THF(30mL)中的反应物混合物SC-MM和SC-NN的溶液(2.1g,6.5mmol)添加BH3.THF(1.0M,13.0mL,13.0mmol),于25℃将溶液搅拌过夜。然后,通过添加水(5mL)淬灭反应。添加2MNaOH溶液(20mL),接着添加30%H2O2(20mL)。在室温将混合物搅拌1小时。用乙酸乙酯(200mL)稀释混合物,并且用卤水(2×100mL)清洗所得的溶液,通过硫酸镁干燥并且在真空中浓缩。在没有进一步纯化的情况下将粗制产物混合物直接用于下一步骤。合成化合物SC-QQ和SC-RR。向二氯甲烷(40mL)中的粗制反应物混合物SC-OO和SC-PP溶液(2.2g,6.5mmol,理论量)按份添加氯铬酸吡啶(Pcc)(2.8g,13.0mmol)。在25℃搅拌溶液过夜。然后,将混合物过滤通过硅胶的短垫并且用二氯甲烷(3×50mL)清洗硅胶。将所有滤液组合并且在真空中浓缩。通过快速层析纯化残留物(石油醚/乙酸乙酯=15:1)以提供产物SC-QQ(910mg,2.7mmol,收率=41%(2步)),为灰白色固体和产物SC-RR(850mg,2.5mmol,收率=39%(2步)),为灰白色固体。化合物SC-QQ:1HNMR(500MHz,CDCl3)δ(ppm):4.17(d,2H),2.53(t,1H),2.17-2.13(m,2H),2.11(s,3H),2.03-2.00(m,1H),0.62(s,3H)。化合物SC-RR:1HNMR(500MHz,CDCl3)δ(ppm):4.45(AB×d,1H),4.39(AB×d,1H),2.54(t,1H),0.62(s,3H)。合成化合物SF。向甲醇(10mL)中的反应物SC-RR溶液(100mg,0.301mmol)添加48%氢溴酸(152mg,0.903mmol),接着是溴(241mg,0.077mL,1.505mmol)。在25℃将溶液加热1.5小时。然后,将混合物倒入冷却水(50mL)中。用乙酸乙酯(2×50mL)提取所得的固体。用卤水(50mL)清洗组合的有机提取物,通过硫酸镁干燥并且在真空中浓缩。在没有进一步纯化的情况下将粗制产物SF直接用于下一步骤。实施例10:合成化合物SF-1。向THF(5mL)中的K2CO3(55mg,0.4mmol)的悬浮液添加2H-四唑(28mg,0.4mmol)和化合物SF(83mg,0.2mmol)。在RT下将混合物搅拌15h,然后将残留混合物倒入5mLH2O并且用EtOAc(2×10mL)提取。用卤水清洗组合的有机层,通过硫酸钠干燥,过滤并且浓缩。通过反相prep-HPLC纯化残留混合物以提供SF-1,为灰白色固体(6mg,8%)。SF-1:1HNMR(500MHz,CDCl3)δ(ppm):8.75(s,1H),5.32(AB,1H),5.19(AB,1H),4.48(AB×d,1H),4.38(AB×d,1H),2.68(t,1H),0.68(s,3H).LC-MS:rt=2.10min,m/z=405.4[M+H]+实施例11:合成化合物SF-2和SF-3.向THF(5mL)中的K2CO3悬浮液(55mg,0.4mmol)添加5-甲基-2H-四唑(33.6mg,0.4mmol)和SF(85mg,0.2mmol),并且在RT将混合物搅拌15h。将残留混合物倒入5mLH2O并且用EtOAc(2×10mL)提取。用卤水清洗组合的有机层,通过硫酸钠干燥,过滤并且浓缩。通过反相prep-HPLC纯化残留混合物以提供SF-2,为灰白色固体(22mg,26%),和SF-3,为灰白色固体(38mg,45%)。SF-2:1HNMR(500MHz,CDCl3)δ(ppm):5.15(AB,1H),5.06(AB,1H),4.48(AB×d,1H),4.39(AB×d,1H),2.68(t,1H),2.47(s,3H),0.69(s,3H).LC-MS:rt=2.09min,m/z=419.3[M+H]+。SF-3:1HNMR(500MHz,CDCl3)δ(ppm):5.35(t,2H),4.48(AB×d,1H),4.38(AB×d,1H),2.63(t,1H),2.56(s,3H),2.25-2.18(m,2H),2.10-2.04(m,1H),0.72(s,3H).LC-MS:rt=2.20min,m/z=419.1[M+H]+实施例12:合成化合物SF-4。向THF(5mL)中的K2CO3悬浮液(55mg,0.4mmol)添加2H-1,2,3-三唑(28mg,0.4mmol)和化合物SF(85mg,0.2mmol)。在RT将混合物搅拌15h,然后将残留混合物倒入5mLH2O并且用EtOAc(2×10mL)提取。用卤水清洗组合的有机层,通过硫酸钠干燥,过滤并且浓缩。通过反相prep-HPLC纯化残留混合物以提供SF-4,为灰白色固体(12mg,15%)。化合物SF-4:1HNMR(500MHz,CDCl3)δ(ppm):7.76(d,1H),7.64(d,1H),5.28(AB,1H),5.14(AB,1H),4.48(AB×d,1H),4.38(AB×d,1H),2.66(t,1H),2.25(s,1H),2.23-2.20(m,1H),2.11-2.08(m,1H),0.68(s,3H).LC-MS:rt=2.05min,m/z=404.3[M+H]+实施例13:合成SG和SG中间体。合成化合物SG-B1和SG-B2。在-78℃在N2下向THF(25mL)和HMPA(0.5mL)中的化合物SC(800mg,2.79mmol)和PhSO2CF2H(540mg,2.79mmol)溶液逐滴添加LHMDS(4mL,THF中的1M)。在–78℃搅拌2h后,用饱和水性NH4Cl溶液(10mL)淬灭反应混合物,并且允许加热到室温然后用Et2O(20mL×3)提取。用卤水清洗组合的有机层,通过硫酸钠干燥,过滤并且浓缩。通过硅胶柱层析(石油醚/乙酸乙酯=10/1)纯化残留物以给出化合物SG-B1和SG-B2的混合物(700mg)。通过手性-HPLC进一步纯化混合物以提供化合物SG-B1(200mg,t=4.31min)。1HNMR(400MHz,CDCl3),δ(ppm),7.99-7.97(d,2H),7.77-7.75(m,1H),7.64-7.60(m,2H),5.14-5.08(m,1H),0.88(s,3H);化合物SG-B2(260mg,t=5.66min)。1HNMR(400MHz,CDCl3),δ(ppm),8.00--7.98(d,2H),7.77-7.75(m,1H),7.64-7.60(m,2H),5.14-5.09(m,1H),0.88(s,3H)。合成化合物SG-C。在–20℃在N2下向无水甲醇(5mL)中的化合物SG-B2(100mg,0.209mmol)和无水Na2HPO4(100mg)溶液添加Na/Hg汞齐(500mg)。在–20℃至0℃搅拌1h后,倾去甲醇溶液,并且用Et2O(5x3mL)清洗固体残留物。用卤水(20mL)清洗组合的有机层,通过MgSO4干燥,过滤并且浓缩。通过硅胶层析(石油醚/乙酸乙酯=10/1)纯化残留物以给出化合物SG-C(36mg,0.106mmol,51%)。1HNMR(400MHz,CDCl3),δ(ppm),6.02-5.88(t,1H),5.17-5.15(m,1H),0.88(s,3H)。合成化合物SG-D。向干THF(5mL)中的化合物SG-C(150mg,0.443mmol)溶液添加硼烷-四氢呋喃复合物(THF中的1.34mL1.0M溶液)。在室温搅拌1小时后,在冰浴中冷却反应混合物,然后用10%水性NaOH(1mL),接着H2O2的30%水溶液(1.2mL)缓慢淬灭。允许混合物在室温搅拌1小时,然后,用EtOAc(3x10mL)提取。用10%水性Na2S2O3(10mL),卤水(10mL)清洗组合的有机层,通过MgSO4干燥,过滤并浓缩以提供粗制化合物SG-D(210mg)。在没有进一步纯化的情况下将粗制产物用于下一步骤。合成化合物SG-E。向10mLH2O饱和二氯甲烷(二氯甲烷已与几毫升H2O一起摇动,然后与水层分离)中溶解的粗制化合物SG-D(210mg)溶液添加Dess-Martin过碘化物(periodinate)(380mg,0.896mmol)。在室温搅拌24h后,用二氯甲烷(3x10mL)提取反应混合物。用10%水性Na2S2O3(10mL),卤水(10mL)清洗组合的有机层,通过MgSO4干燥,过滤并且浓缩。通过硅胶上层析(石油醚/乙酸乙酯=5:1)纯化残留物以提供化合物SG-E(90mg,0.254mmol,57%),为灰白色固体。1HNMR(400MHz,CDCl3),δ(ppm),6.01-5.73(t,1H),2.55-2.54(m),2.12(s),0.62(s,3H)。合成化合物SG。向MeOH(5mL)中的化合物SG-E(80mg,0.226mmol)的溶液添加2滴HBr(48%),接着添加溴(100mg,0.63mmol)。在室温搅拌1h后,将反应混合物倒入冰水中,然后用乙酸乙酯(15mLx3)提取,用卤水(20mL)清洗组合的有机层,通过MgSO4干燥,过滤并浓缩以给出粗制化合物SG(95mg)。在没有进一步纯化的情况下将粗制产物用于下一步骤。实施例14:合成化合物SG-1和SG-2。向THF(5mL)中的K2CO3(55mg,0.4mmol)悬浮液添加2H-四唑(28mg,0.4mmol)和10(86mg,0.2mmol)。在RT将混合物搅拌15h。将残留混合物倒入5mLH2O并且用EtOAc(2×10mL)提取。用卤水清洗组合的有机层,通过硫酸钠干燥,过滤并且浓缩。通过反相prep-HPLC纯化残留混合物以提供SG-1,为灰白色固体(12mg,14.2%)和灰白色固体副产物(15mg,17.7%)。SG-1:1HNMR(500MHz,CDCl3)δ(ppm):8.74(s,1H),5.87(t,1H),5.32(AB,1H,J=18.0Hz),5.19(AB,1H),2.68(t,1H,J=8.5Hz),2.26-2.20(m),2.09-2.05(m),0.68(s,3H).LC-MS:rt=2.11min,m/z=423.3[M+H]+实施例15:合成化合物SG-3和SG-4。向THF(5mL)中的K2CO3(55mg,0.4mmol)悬浮液添加5-甲基-2H-四唑(28mg,0.4mmol)和SG(86mg,0.2mmol)。在RT将混合物搅拌15h。将残留混合物倒入5mLH2O并且用EtOAc(2×10mL)提取。用卤水清洗组合的有机层,通过硫酸钠干燥,过滤并且浓缩。通过反相prep-HPLC纯化残留混合物以提供SG-3,为灰白色固体(15mg,17%),和SG-4,为灰白色固体(30mg,34%)。SG-3:1HNMR(500MHz,CDCl3)δ(ppm):5.87(t,1H),5.15(AB,1H),5.05(AB,1H),2.67(t,1H),2.47(s,3H),2.22-2.20(m,1H)2.09-2.07(m,1H),0.69(s,3H).LC-MS:rt=2.14min,m/z=437.1[M+H]+.SG-4:1HNMR(500MHz,CDCl3)δ(ppm):5.87(t,1H),5.35(s,2H),2.63(t,1H),2.56(s,3H),0.72(s,3H).LC-MS:rt=2.24min,m/z=437.0[M+H]+实施例16:合成化合物SG-5。向THF(5mL)中的K2CO3(25mg,0.18mmol)悬浮液添加1H-1,2,3-三唑(50mg,0.72mmol)和反应物(100mg,0.23mmol)。在室温将混合物搅拌15h,然后,将反应混合物倒入10mLH2O中并且用EtOAc(2×20mL)提取。用卤水(10mL)清洗组合的有机层,通过硫酸钠干燥,过滤并且在真空中浓缩。通过反相prep-HPLC纯化残留混合物以提供标题化合物SG-5(15.4mg,0.0365mmol,22%)。SG-5:1HNMR(400MHz,CDCl3)δ(ppm):7.75(s,1H),7.64(s,1H),5.87(t,1H),5.27(AB,1H),5.14(AB,1H),2.66(t,1H),0.69(s,3H)。实施例17:合成SE和SE中间体。合成化合物SE-A。经由注射泵在30min里在0℃向THF(20mL)中的EtMgBr(THF中的5mmol,1M)溶液添加干THF(5mL)中的化合物SC(858mg,3mmol)溶液。在0℃搅拌5h后,允许反应混合物加热,并且在室温搅拌过夜。用冰冷的水淬灭反应混合物,并且用EtOAc(15mLx3)提取。用卤水清洗组合的有机层,通过硫酸钠干燥,过滤并且浓缩。通过快速柱层析(石油醚/乙酸乙酯=20:1至10:1)纯化白色残留物以给出化合物SE-A(900mg)。合成化合物SE-B。向干THF(5mL)中的化合物SE-A(200mg,0.66mmol)溶液添加硼烷-四氢呋喃复合物(2mL在THF中的1.0M溶液)。在室温搅拌1小时后,在冰浴中冷却反应混合物,然后用10%水性NaOH(1mL),接着用H2O2的30%水溶液(1.2mL)缓慢淬灭。允许混合物在室温搅拌1小时然后,用EtOAc(3x10mL)提取。用10%水性Na2S2O3(10mL),卤水(10mL)清洗组合的有机层,通过MgSO4干燥,过滤并浓缩以提供化合物SE-B(260mg,粗制)。在没有进一步纯化的情况下将粗制产物用于下一步骤。合成化合物SE-C。向10mL二氯甲烷中溶解的化合物SE-B(260mg,粗制)的溶液添加PCC(449mg,)。在室温搅拌24h后,用二氯甲烷(3x10mL)提取反应混合物。用10%水性NaCl(10mL),卤水(10mL)清洗组合的有机层,通过MgSO4干燥,过滤并且浓缩。通过硅胶上层析(石油醚/乙酸乙酯=4:1至2:1)纯化残留物以提供标题SE-C(15mg,),为灰白色固体。1HNMR(500MHz,CDCl3),δ(ppm),2.49(1H,t),0.84(,t3H),0.59(s,3H)。合成化合物SE。向MeOH(5mL)中的化合物SE-C(30mg,0.09mmol)溶液添加2滴HBr(48%),接着是溴(100mg,0.62mmol)。在室温搅拌1h后,将反应混合物倒入冰水中,然后用乙酸乙酯(15mLx3)提取,用卤水清洗组合的有机层(20mL),通过MgSO4干燥,过滤并浓缩以给出化合物SE(36mg粗制)。在没有进一步纯化的情况下将粗制产物用于下一步骤。实施例18:合成化合物SE-1和SE-2。向THF(5mL)中的K2CO3(50mg,0.36mmol)悬浮液添加1H-四唑(40mg,0.46mmol)和SM(100mg,0.243mmol)。在rt将混合物搅拌15h。将反应混合物倒入5mLH2O并且用EtOAc(2×10mL)提取。用卤水清洗组合的有机层,通过硫酸钠干燥,过滤并且浓缩。通过反相prep-HPLC纯化残留物以提供标题化合物,为灰白色固体SE-1(9mg,9.2%),SE-2(15mg,15.6%)。SE-1:1HNMR(400MHz,CDCl3)δ(ppm):8.75(s,1H),5.32(AB,1H),5.20(AB,1H),2.67(t,1H),1.59(q,2H),0.88(t,3H),0.68(s,3H)。LC-MS:rt=2.27min,m/z=383.4(M+-H2O+1).SE-2:1HNMR(400MHz,CDCl3),δ(ppm):8.57(s,1H),5.46(s,2H),2.67(t,1H),1.59(q,2H),0.88(t,3H),,0.71(s,3H).LC-MS:rt=2.36min,m/z=383.4(M+-H2O+1)。实施例19:合成化合物SE-3和SE-4。向THF(5mL)中的K2CO3(50mg,0.36mmol)悬浮液添加2H-1,2,3-三唑(36mg,0.52mmol)和SE(100mg,0.25mmol)。在rt搅拌混合物24小时。然后,将反应混合物倒入5mLH2O中并且用EtOAc(2×10mL)提取。用卤水清洗组合的有机层,通过硫酸钠干燥,过滤并且浓缩。通过反相prep-HPLC纯化残留物以提供标题化合物,为灰白色固体,SE-3(9mg,9.3%),SE-4(10mg,10.3%),SE-3:1HNMR(500MHz,CDCl3)δ(ppm):7.75(d,1H),7.64(d,1H),5.27(AB,1H),5.13(AB,1H),2.67(1H,t),1.59(2H,q),0.90(3H,t),1.28(s,3H),0.67(s,3H)。LC-MS:rt=2.31min,m/z=400.4(M++1).SE-4:1HNMR(500MHz,CDCl3)δ(ppm):7.68(s,2H),5.25(AB,1H),5.21(AB,1H),2.58(t,1H),1.59(2H,q),0.90(3H,t),0.71(s,3H).LC-MS:rt=2.42min,m/z=400.4(M++1)。实施例20:合成化合物SE-5和SE-6。向THF(5mL)中的K2CO3(55mg,0.4mmol)悬浮液添加5-甲基-2H-四唑(33.6mg,0.4mmol)和化合物SE(82mg,0.2mmol)。在RT将混合物搅拌15hthen将残留混合物倒入5mLH2O并且用EtOAc(2×10mL)提取。用卤水清洗组合的有机层,通过硫酸钠干燥,过滤并且浓缩。通过反相prep-HPLC纯化残留混合物以提供SE-5,为灰白色固体(11.1mg,13.5%),和SE-6,为灰白色固体(30.6mg,37.2%)。SE-5:1HNMR(500MHz,CDCl3)δ(ppm):5.13(AB,1H),5.07(AB,1H),2.66(t,1H),2.47(s,3H),2.24-2.17(m,1H),2.11-2.05(m,1H),1.47(q,2H),0.93(t,3H),0.69(s,3H).LC-MS:rt=2.13min,m/z=415.1[M+H]+.SE-6:1HNMR(500MHz,CDCl3)δ(ppm):5.37(AB,1H),5.33(AB,1H),2.62(t,1H),2.56(s,3H),2.25-2.18(m,1H),2.09-2.06(m,1H),1.47(q,2H),0.93(t,3H),0.72(s,3H).LC-MS:rt=2.26min,m/z=415.3[M+H]+实施例21:合成SM和SM中间体。合成化合物SA-DD和SA-EE。在干甲醇(250mL)中溶解化合物混合物SA-BB和SA-CC(5.0g,16.7mmol),并且添加Na金属(1.2g,50.0mmol),并且将溶液回流16h。蒸发掉甲醇,并且在二氯甲烷中溶解残留物,并且用H2O(3x50mL)和卤水(100mL)清洗,通过MgSO4干燥,过滤,并且浓缩。通过硅胶层析(石油醚/乙酸乙酯=10:1至5:1)纯化粗制靶标化合物,并且浓缩以给出产物混合物SA-DD和SA-EE(4.6g,83%),为灰白色固体。合成化合物SA-FF和SA-GG。向无水THF(30mL)中的反应物混合物SA-DD和SA-EE(4.6g,13.9mmol)溶液添加BH3.THF(1.0M,27.7mL,27.7mmol),在25℃将溶液搅拌过夜,然后,通过添加水(5mL)淬灭反应。添加2MNaOH溶液(30mL),接着添加30%H2O2(30mL)。在室温将混合物搅拌1小时。用乙酸乙酯(200mL)稀释混合物,并且用卤水(2×100mL)清洗所得的溶液,通过硫酸镁干燥并且在真空中浓缩。在没有进一步纯化的情况下将粗制产物混合物直接用于下一步骤。合成化合物SA-HH和SA-II。向二氯甲烷(40mL)中的粗制反应物混合物SA-FF和SA-GG(4.9g,13.9mmol,理论量)溶液按份添加氯铬酸吡啶(PCC)(6.0g,27.8mmol)。在25℃将溶液搅拌过夜,然后将混合物过滤通过硅胶的短垫并且用二氯甲烷(3×50mL)清洗硅胶。组合所有滤液,并且在真空中浓缩。通过快速层析纯化残留物(石油醚/乙酸乙酯=15:1)以提供产物SA-HH(2.1g,6.03mmol,收率=43%(2步)),为灰白色固体,和产物SA-II(2.2g,6.32mmol,收率=45%(2步)),为灰白色固体。化合物SA-HH:1HNMR(500MHz,CDCl3)δ(ppm):3.40(s,3H),3.20(s,2H),2.62-2.51(m,2H),2.11(s,3H),2.02-1.99(m,2H),0.62(s,3H)。化合物SA-II:1HNMR(500MHz,CDCl3)δ(ppm):3.42(AB,1H),3.38(AB,1H),3.40(s,3H),2.65(s,1H),2.54(t,1H),2.16-2.14(m,1H),2.11(s,3H),2.02-1.98(m,1H),0.61(s,3H)。合成化合物SM。向甲醇(10mL)中的反应物SA-II(100mg,0.301mmol)溶液添加48%氢溴酸(152mg,0.903mmol),接着是溴(241mg,0.077mL,1.51mmol)。在25℃将溶液加热1.5小时,然后,将混合物倒入冷水(50mL),并且用乙酸乙酯(2×50mL)提取所得的固体。用卤水(50mL)清洗组合的有机提取物,通过硫酸镁干燥并且在真空中浓缩。在没有进一步纯化的情况下将粗制产物SM直接用于下一步骤。实施例22:合成化合物SM-1。向THF(3mL)中的化合物SM(120mg,0.28mmol)溶液添加K2CO3(190mg,1.4mmol)和1H-四唑(100mg,1.4mmol)。在室温将所得的溶液搅拌过夜,然后用EtOAc(20mL)稀释反应。用卤水(10mL)清洗所得的溶液,通过Na2SO4干燥并且在真空中浓缩。通过prep-HPLC纯化残留物以给出SM-1(12mg,10%),和灰白色固体副产物(14mg,12%)。SM-1:1HNMR:(500MHz,CDCl3),δ(ppm),8.74(s,1H),5.32(AB,1H),5.19(AB,1H),3.42(AB,1H),3.40(S,3H),3.39(AB,1H),2.68(t,1H),2.66(s,1H),0.67(s,3H).LC-MS:rt=2.19min;m/z=399.2(M-18)+实施例23:合成化合物SM-3和SM-4。向THF(5mL)中的K2CO3(55mg,0.4mmol)悬浮液添加5-甲基-2H-四唑(33.6mg,0.4mmol)和10(85mg,0.2mmol)。在RT将混合物搅拌15h,然后倒入5mLH2O并且用EtOAc(2×10mL)提取。用卤水清洗组合的有机层,通过硫酸钠干燥,过滤并且浓缩。通过反相prep-HPLC纯化残留混合物以提供SM-3,为灰白色固体(8.6mg,10%)和灰白色固体(12mg,13.9%)。SM-3:1HNMR(500MHz,CDCl3)δ(ppm):5.15(AB,1H),5.05(AB,1H),3.42(AB,1H),3.39(AB,1H),3.40(s,3H),2.67(t,1H),2.64(s,1H),2.47(s,3H),2.21-2.17(m,1H),2.08-2.05(m,1H),0.68(s,3H).LC-MS:rt=2.14min,m/z=431.2[M+H]+.SM-4:1HNMR(500MHz,CDCl3)δ(ppm):5.37(AB,1H),5.33(AB,1H),3.42(AB,1H),3.38(AB,1H),3.40(s,3H),2.63(t,1H),2.56(s,3H),0.71(s,3H).LC-MS:rt=2.25min,m/z=431.2[M+H]+实施例24:合成化合物SM-5。向THF(5mL)中的K2CO3(55mg,0.4mmol)悬浮液添加2H-1,2,3-三唑(28mg,0.4mmol)和化合物SM(85mg,0.2mmol)。在RT将混合物搅拌15h,然后将残留混合物倒入5mLH2O中并且用EtOAc(2×10mL)提取。用卤水清洗组合的有机层,通过硫酸钠干燥,过滤并且浓缩。通过反相prep-HPLC纯化残留混合物以提供SM-5,为灰白色固体(25mg,30%)。化合物SM-5:1HNMR(500MHz,CDCl3)δ(ppm):7.76(s,1H),7.65(s,1H),5.28(AB,1H),5.14(AB,1H),3.42(AB,1H),3.39(AB,1H),3.40(s,3H),2.66(t,1H),2.23-2.20(m,1H),2.10-2.08(m,1H),0.67(s,3H).LC-MS:rt=2.14min,m/z=415.8[M+H]+实施例25:合成SO和SO中间体。合成化合物SO-C和SO-D。在干乙醇(250mL)中溶解化合物混合物SO-A和SO-B(5.0g,16.7mmol),并且添加Na(1.2g,50.0mmol)。将溶液回流16h。蒸发掉乙醇,并且在二氯甲烷中溶解残留物,并且用H2O(3x50mL)和卤水(100mL)清洗,通过MgSO4干燥,过滤,并且浓缩。通过硅胶层析(石油醚/乙酸乙酯=10:1至5:1)纯化粗制靶标化合物,并且浓缩以给出产物混合物SO-C和SO-D(4.5g,78%),为灰白色固体。合成化合物SO-E和SO-F。向无水THF(30mL)中的反应物混合物SO-C和SO-D(4.5g,13.0mmol)溶液添加BH3.THF(1.0M,27.7mL,27.7mmol),在25℃搅拌溶液过夜。然后,通过添加水(5mL)淬灭反应。添加2MNaOH溶液(30mL),接着添加30%H2O2(30mL)。在室温将混合物搅拌1小时。用乙酸乙酯(200mL)稀释混合物,并且用卤水(2×100mL)清洗所得的溶液,通过硫酸镁干燥并且在真空中浓缩。在没有进一步纯化的情况下将粗制产物混合物直接用于下一步骤。合成化合物SO-G和SO-H。向二氯甲烷(40mL)中的粗制反应物混合物SO-E和SO-F(4.5g,13.0mmol,理论量)溶液按份添加氯铬酸吡啶(PCC)(5.7g,26.0mmol)。在25℃搅拌溶液过夜。然后,将混合物过滤通过硅胶的短垫并且用二氯甲烷(3×50mL)清洗硅胶。将所有滤液组合并且在真空中浓缩。通过快速层析纯化残留物(洗脱剂:石油醚/乙酸乙酯=15:1)以提供产物SO-G(2.0g,5.5mmol,收率=42%(2步)),为灰白色固体,和产物SO-H(1.8g,4.97mmol,收率=38%(2步)),为灰白色固体。SO-H:1HNMR(500MHz,CDCl3)δ(ppm):3.53(q,2H),3.45(AB,1H),3.41(AB,1H),2.54(t,1H),2.16-2.12(m),2.11(s),2.02-1.98(m),1.2(t,3H),0.61(s,3H)。合成化合物SO。向甲醇(10mL)中的反应物SO-H(100mg,0.301mmol)溶液添加48%氢溴酸(152mg,0.903mmol),接着是溴(241mg,0.077mL,1.505mmol)。在25℃将溶液加热1.5小时。然后,将混合物倒入冷却水(50mL)。用乙酸乙酯(2×50mL)提取所得的固体。用卤水(50mL)清洗组合的有机提取物,通过硫酸镁干燥并且在真空中浓缩。在没有进一步纯化的情况下将粗制产物SO直接用于下一步骤。实施例26:合成化合物SO-1和SO-2。向THF(5mL)中的K2CO3(55mg,0.4mmol)悬浮液添加5-甲基-2H-四唑(33.6mg,0.4mmol)和10(85mg,0.2mmol)。在rt将混合物搅拌15h。将残留混合物倒入5mLH2O中并且用EtOAc(2×10mL)提取。用卤水清洗组合的有机层,通过硫酸钠干燥,过滤并且浓缩。通过反相prep-HPLC纯化残留混合物以提供SO-1,为灰白色固体(9.6mg,10.8%),和SO-2,为灰白色固体(17.5mg,19.7%)。SO-1:1HNMR(500MHz,CDCl3)δ(ppm):5.15(AB,1H),5.05(AB,1H),3.54(q,2H),3.45(AB,1H),3.41(AB,1H),2.75(s,1H),2.66(t,1H),2.47(s,3H),2.24-2.17(m,1H),2.08-2.05(m,1H),1.21(t,3H),0.68(s,3H).LC-MS:rt=2.24min,m/z=445.3[M+H]+。SO-2:1HNMR(500MHz,CDCl3)δ(ppm):5.36(AB,1H),5.35(AB,1H),3.54(q,2H),3.45(AB,1H),3.41(AB,1H),2.75(s,1H),2.63(t,1H),2.56(s,3H),2.24-2.17(m,1H),2.09-2.05(m,1H),1.21(t,3H),0.71(s,3H).LC-MS:rt=2.35min,m/z=427.3[M-H2O+H]+实施例27:合成SL和SL中间体。合成化合物SL-B。将SA-A(10g,36.7mmol)添加到50mL氯化乙酰和50mlL乙酸酐。将反应混合物加热到120℃达5h,在真空中蒸发以提供粗制SL-B,为灰白色固体(10g,87%收率)。1HNMR(400MHz,CDCl3),δ(ppm),5.78(s,1H),5.55(s,1H),2.4(2H,dd),2.13(s,3H),0.90(s,3H).合成化合物SL-C。在0℃向200mLTHF和20mLH2O中的SL-B(10g,31.8mmol)溶液,添加mCPBA(11g,63.6mmol),在rt搅拌15h,500mLEtOAc提取反应混合物,用100mL饱和Na2SO3,100mL饱和NaHCO3和100mL卤水清洗,并且在真空中蒸发,然后通过层析(PE:EtOAc=5:1)纯化以提供SL-C,为灰白色固体(2.2g,24%收率)。1HNMR(400MHz,CDCl3),δ(ppm),5.92(s,1H),4.44(s,1H),0.95(s,3H)。合成化合物SL-D。向50mLEtOAc中的SL-C(2g,6.94mmol)溶液,添加Pd/C200mg。在1atmH2将反应混合物氢化15h。在真空中蒸发反应混合物,然后通过层析(PE:EtOAc=1:2)纯化以提供SL-D,为灰白色固体(1g,50%收率)。1HNMR(400MHz,CDCl3),δ(ppm),3.83(s,1H),0.93(s,3H)。合成化合物SL-E。向100mLMeOH中的SL-D(1g,3.4mmol)溶液,添加TsOH50mg,加热到60℃达2h。用500mLEtOAc提取反应混合物,用100mL饱和NaHCO3,100mL卤水清洗并且在真空中蒸发以提供SL-E,为灰白色固体(1g,91%收率)。1HNMR(400MHz,MeOD),δ(ppm),3.80(s,1H),3.20(s,3H),3.15(s,3H),0.89(s,3H)。合成化合物SL-F。向30mLTHF中的乙基三苯基溴化膦(ethyltriphenylphosphoniumbromide)(10.67g,28.84mmol)溶液,添加KOt-Bu(3.23g,28.80mmol)。将反应加热到60℃达1h,然后将SL-E(3.23g,9.6mmol)添加到混合物,在60℃搅拌15h。用500mLEtOAc提取反应混合物,用卤水清洗,并且在真空中蒸发,然后通过层析(PE:EtOAc=3:1)纯化,以提供SL-F,为灰白色固体(2.18g,65%收率)。1HNMR(400MHz,d6-丙酮),δ(ppm),5.09-5.07(m,1H),3.65(s,1H),3.11(s,3H),3.08(s,3H),0.88(s,3H)。合成化合物SL-G。向50mLTHF中的SL-F(1g,2.9mmol)溶液,添加NaH(2g,5.8mmol),在rt搅拌1h。然后,将1mLMeI添加到混合物,在rt搅拌过夜。用5mLH2O淬灭反应混合物,并且用100mLEtOAc提取,用卤水清洗并且在真空中蒸发,然后通过层析(PE:EtOAc=10:1)纯化以提供SL-G,为灰白色固体(577mg,55%收率)。.1HNMR(400MHz,d6-丙酮),δ(ppm),4.96-4.93(m,1H),3.12(s,3H),3.00(s,1H),2.98(s,3H),2.96(s,3H),0.75(s,3H)。合成化合物SL-H。向20mLTHF中的SL-G(1g,2.8mmol)溶液,添加2M含水HCl2mL,在rt搅拌1h。用5mLH2O淬灭反应混合物,并且用100mLEtOAc提取,用卤水清洗并且在真空中蒸发然后通过层析纯化(PE:EtOAc=10:1)以提供SL-H,作为灰白色固体(750mg,83%收率)。1HNMR(400MHz,CDCl3),δ(ppm),5.15-5.11(m,1H),3.32(s,3H),3.14(s,1H),0.92(s,3H)。合成化合物SL-I。向10mLDMSO中的三甲基碘化锍(6.4g,31.5mmol)的搅拌溶液添加NaH(60%,800mg,31.5mmol)。在室温搅拌1h后,逐滴添加5mLDMSO中的SL-H(1g,3.2mmol)的悬浮液。在15h后,将反应混合物倒入冰冷的水中,并且用300mLEtOAc提取,用100mL卤水清洗,干燥并且在真空中蒸发然后通过层析纯化(PE:EtOAc=10:1)以提供SL-I及其异构体,为灰白色固体(793mg,76%收率)。合成化合物SL-J。向10mLTHF中的SL-I及其异构体(150mg,0.45mmol)溶液,添加LiAH4(50mg,1.35mmol),在rt搅拌1h。用5mLH2O淬灭反应混合物,并且用100mLEtOAc提取,用卤水清洗并且在真空中蒸发然后通过层析纯化(PE:EA=3:1)以提供SL-J,为灰白色固体(72mg,48%收率)。1HNMR(400MHz,CDCl3),δ(ppm),5.11-5.10(m,1H),3.33(s,3H),3.12(s,1H),1.22(s,3H),0.89(s,3H)。合成化合物SL-K。向干THF(5mL)中的SL-J(100mg,0.3mmol)溶液添加硼烷-四氢呋喃复合物(1mL;THF中的1.0M溶液)。在室温搅拌1小时后,在冰浴中冷却反应混合物,然后用10%水性NaOH(1mL)接着用30%H2O2的水溶液(1mL)缓慢淬灭。在室温搅拌1小时后,用EtOAc(3x100mL)提取混合物。用10%水性Na2S2O3(100mL),卤水(100mL)清洗组合的有机层,通过MgSO4干燥,过滤并浓缩以提供SL-K,为灰白色固体(100mg,91%)。在没有进一步纯化的情况下将粗制产物用于下一步骤。合成化合物SL-L。向20mLDCM中的SL-K(100mg,0.29mmol)溶液,添加PCC(190mg,0.87mmol),在rt搅拌2h。用5mLH2O淬灭反应混合物,并且用100mLEtOAc提取,用卤水清洗并且在真空中蒸发然后通过层析纯化(PE:EtOAc=3:1)以提供SL-L,为灰白色固体(55mg,55%收率)。1HNMR(400MHz,CDCl3),δ(ppm),3.30(s,3H),3.10(s,1H),2.5(1H,t,J=10Hz),2.1(s,3H),1.16(s,3H),0.56(s,3H)。合成化合物SL。向MeOH(5mL)中的SL-L(40mg,0.11mmol)溶液添加2滴HBr(48%),接着是溴(150mg,0.33mmol)。在室温搅拌1小时后,将反应混合物倒入冰水中,然后用EtOAc(10mLx3)提取。用卤水(20mL)清洗组合的有机层,通过MgSO4干燥,过滤并浓缩以给出粗制化合物SL,为灰白色固体(40mg,80%收率)。在没有进一步纯化的情况下将粗制产物用于下一步骤。实施例28:合成化合物SL-1和SL-2。向THF(5mL)中的SL(40mg,0.09mmol)悬浮液添加1H-1,2,3-三唑(30mg,0.45mmol)和K2CO3(60mg,0.45mmol)。在25℃将混合物搅拌15h。通过反相prep-HPLC纯化反应混合物以提供SL-1,为灰白色固体(5mg,13%收率),和SL-2,为灰白色固体(5mg,13%收率)。SL-1:1HNMR(400MHz,CDCl3),δ(ppm),7.75(s,1H),7.64(s,1H),5.25-5.13(m,2H),3.31(s,3H),3.11(s,1H),1.24(s,3H),0.71(s,3H).SL-2:1HNMR(400MHz,CDCl3),δ(ppm),7.68(s,2H),5.27-5.19(m,2H),3.31(s,3H),3.11(s,1H),1.21(s,3H),0.75(s,3H)。实施例29:合成SH和SH中间体。合成化合物SH-C。在0℃向200mLTHF和20mLH2O中的化合物SL-B(10g,31.8mmol)溶液添加m-CPBA(11g,63.6mmol)。在rt搅拌15h后,用500mLEtOAc稀释反应混合物。用300mL饱和Na2SO3,300mL饱和NaHCO3和300mL卤水清洗所得的溶液,并且在真空中蒸发。通过层析(PE:EA=5:1)纯化残留物以提供SH-C,为灰白色固体(1.1g,3.8mmol,12%收率)。1HNMR(500MHz,CDCl3),δ(ppm),6.25(s,1H),4.27(dd,1H),0.93(s,3H)。合成化合物SH-D。向50mLEtOAc中的化合物SH-C(2g,6.94mmol)溶液添加Pd\C200mg。在1atmH2中氢化反应混合物15h。将反应混合物在真空中蒸发然后通过层析纯化(PE:EA=1:2)以提供SH-D,为灰白色固体(1.5g,5.2mmol,75%收率)。1HNMR(500MHz,CDCl3),δ(ppm),3.97(td,1H),0.88(s,3H)。合成化合物SH-E。向100mLMeOH中的化合物SH-D(1g,3.4mmol)溶液,添加TsOH50mg。将溶液加热到60℃达2h。然后,用500mLEtOAc稀释反应混合物,用100mL饱和NaHCO3,100mL卤水清洗,并且在真空中蒸发以提供SH-E,为灰白色固体(1g,91%收率)。合成化合物SH-F。向30mLTHF中的乙基三苯基溴化膦(10.67g,28.84mmol)溶液添加KOt-Bu(3.23g,28.80mmol)。将反应加热到60℃达1h,然后将化合物SH-E(3.23g,9.6mmol)添加到混合物。于60℃将溶液加热15h。然后,用500mLEtOAc稀释反应混合物。将所得的溶液用100mL卤水清洗,在真空中蒸发,然后通过层析纯化(PE:EA=3:1)以提供SH-F,为灰白色固体(2g,5.74mmol,62%收率)。1HNMR(500MHz,MeOD),δ(ppm),5.15-5.12(m,1H),3.80-3.78(m,1H),3.21(s,3H),3.15(s,3H),1.67(d,3H),0.95(s,3H)。合成化合物SH-G。在-78℃向10mLDCM中的化合物SH-F(0.5g,1.43mmol)溶液添加DAST(0.5ml,10mmol)。在-78℃将反应混合物搅拌30min,然后用5Ll饱和NaHCO3淬灭,用50mlDCM提取,用100mL卤水清洗,通过Na2SO4干燥,在真空中浓缩,并且通过层析(PE:EA=30:1)纯化以提供SH-G,为灰白色固体(175mg,0.5mmol,35%收率)。合成化合物SH-H。向20mLTHF中的化合物SH-G(350mg,1mmol)溶液添加2MHCl(2mL)。在rt搅拌溶液1h,然后用100mLEtOAc提取反应混合物,用100mL卤水清洗,并且在真空中蒸发。然后通过层析(PE:EA=10:1)纯化所得的残留物以提供SH-H,为灰白色固体(210mg,0.7mmol,60%收率)。1HNMR(500MHz,CDCl3),δ(ppm),5.17-5.14(m,1H),4.80-4.66(m,1H),2.61-2.57(m,1H),1.79(d,3H),0.93(s,3H)。合成化合物SH-I。向10mLDMSO中的三甲基碘化锍(3.2g,16mmol)的搅拌悬浮液添加NaH(60%,400mg,16mmol)。在室温搅拌1h后,逐滴添加5mLDMSO中的化合物SH-H(486mg,1.6mmol)悬浮液。在15h后,将反应混合物倒入冰冷的水中,并且用300mLEtOAc提取。用100mL卤水清洗所得的溶液,干燥(NaSO4),并且在真空中蒸发。然后通过层析(PE:EA=10:1)纯化所得的残留物以提供SH-I及其C-3异构体的混合物,为灰白色固体(290mg,0.91mmol,58%收率)。合成化合物SH-J。向10mlTHF中的SH-I及其C-3异构体(300mg,0.94mmol)溶液,添加LiAH4(100mg,2.7mmol)。将悬浮液在rt搅拌1h。然后,用5mLH2O淬灭反应混合物,并且用100mLEtOAc提取。将所得的溶液用卤水清洗并且在真空中蒸发。然后通过层析纯化所得的残留物(PE:EA=3:1)以提供SH-J,为灰白色固体(140mg,48%收率)。1HNMR(500MHz,CDCl3),δ(ppm),5.15-5.12(m,1H),4.72-4.60(m,1H),1.70(d,3H),1.27(s,3H),0.92(s,3H)。合成化合物SH-K。向干THF(5mL)中的化合物SH-J(100mg,0.3mmol)溶液添加硼烷-四氢呋喃复合物(1mL;THF中的1.0M溶液)。在室温搅拌1小时后,在冰浴中冷却反应混合物,然后用10%水性NaOH(1mL),接着用H2O2的30%水溶液(1mL)缓慢淬灭。在室温搅拌1小时后,用EtOAc(3x100mL)提取混合物。然后,用10%水性Na2S2O3(100mL),卤水(100mL)清洗组合有机提取物,通过MgSO4干燥,过滤并浓缩以提供粗制SH-K,为灰白色固体(100mg,91%)。在没有进一步纯化的情况下将粗制产物用于下一步骤。合成化合物SH-L。向20mDCLM中的化合物SH-K(100mg,0.29mmol)溶液添加PCC(190mg,0.87mmol),并且将所得的溶液在rt搅拌2h。然后,将反应混合物过滤通过铈硅石的垫,并且在真空中蒸发滤液。然后通过层析(PE:EA=3:1)纯化残留物以提供SH-L,为灰白色固体(53mg,53%收率)。1HNMR(400MHz,CDCl3),δ(ppm),4.71-4.57(m,1H),2.54(1H,t),2.15(s,3H),1.28(s,3H),0.58(s,3H)。合成化合物SH。向MeOH(5mL)中的化合物SH-L(40mg,0.11mmol)溶液添加2滴HBr(48%),接着是溴(150mg,0.33mmol)。在室温搅拌1h后,将反应混合物倒入冰水中,然后用乙酸乙酯(10mLx3)提取。用卤水清洗组合的有机层(20mL),通过MgSO4干燥,过滤并浓缩以给出粗制化合物SH,为黄色固体(40mg,80%收率)。在没有进一步纯化的情况下将粗制产物用于下一步骤。实施例30:合成化合物SH-1和SH-2。向THF(5mL)中的化合物SH(50mg,0.12mmol)悬浮液添加2H-1,2,3-三唑(120mg,1.8mmol)和K2CO3(200mg,1.2mmol)。在25℃将混合物搅拌15h。用乙酸乙酯(20mLx3)提取反应混合物。用卤水(20mL)清洗组合的有机层,通过MgSO4干燥,过滤并浓缩以给出粗制产物。通过反相prep-HPLC纯化此粗制产物以提供SH-1,为灰白色固体(12mg,0.03mmol,25%收率),和SH-2,为灰白色固体(5.7mg,0.014mmol,8.33%收率)。SH-1:1HNMR(500MHz,CDCl3),δ(ppm),7.76(s,1H),7.65(s,1H),5.29(1H,AB),5.14(1H,AB),4.73-4.59(m,1H),2.68(1H,t),1.30(s,3H),0.67(s,3H).SH-2:1HNMR(500MHz,CDCl3),δ(ppm),7.69(s,2H),5.27(1H,AB),5.23(1H,AB),4.73-4.59(m,1H),4.64-4.59(m,1H),2.60(1H,t),1.29(s,3H),0.70(s,3H)。实施例31:合成SB和SB中间体合成化合物SB-B和SB-C。在三颈烧瓶中,在-70℃,将小块的锂(7.63g,1.1mol)加入到2.7升冷凝氨中。一旦所有的锂溶解,将蓝色溶液加热至-50℃。逐滴加入19-去甲雄-4-烯-3,17-二酮SB-A(1,30g,110mmol)和tert-BuOH(8.14g,110mmol)的800mL无水四氢呋喃溶液,搅拌90分钟,直到反应混合物变成浅黄色为止。加入氯化铵(70g),并蒸发过量的氨。将残余物用0.5NHCl(500mL)和二氯甲烷(500mLx2)提取。将组合的有机层用饱和NaHCO3溶液洗涤,用Na2SO4干燥,过滤,浓缩,得到SB-B和SB-C的混合物(21g,70%),其不用进一步纯化,在下一步中直接使用。将SB-B和SB-C(21g,76mmol)的50mL无水二氯甲烷溶液加入到氯铬酸吡啶(PCC)(32.8g,152mmol)的450mL二氯甲烷悬浮液中。在室温搅拌2小时之后,将2NNaOH溶液(500mL)加入到暗褐色反应混合物中,并再搅拌10分钟。将得到的溶液用二氯甲烷提取,用2NHCl、卤水洗涤组合的有机层,用Na2SO4干燥,过滤,浓缩。用硅胶色谱纯化残余物(石油醚/乙酸乙酯=20:1至10:1),提供标题化合物SB-C(16.8g,80%)的灰白色固体。SB-B的1HNMR(400MHz,CDCl3)δ(ppm),3.65(t,1H,1H),0.77(s,3H)。SB-C的1HNMR(400MHz,CDCl3)δ(ppm),0.88(s,3H)。合成化合物SB-D.向化合物SB-C(16.8g,61.3mmol)的甲醇(250mL)溶液中加入碘(1.54g,6.1mmol)。在60℃搅拌12小时之后,真空除去溶剂。将粗品溶于二氯甲烷(200mL)中,并用饱和NaHCO3(150mL)、卤水洗涤,用Na2SO4干燥,过滤,浓缩。用碱性氧化铝色谱纯化残余物(石油醚/乙酸乙酯=100:1),得到化合物SB-D(14g,43.8mmol,71%)。1HNMR(400MHz,CDCl3)δ(ppm),3.18(s,3H),3.12(s,3H),0.85(s,3H)。合成化合物SB-E.在0℃,向t-BuOK(7.36g,65.7mmol)的THF(100mL)悬浮液中慢慢地加入乙基三苯基溴化鏻(26g,70mmol)。在60℃搅拌3小时之后,加入化合物SB-D(7g,21.9mmol),并将该混合物在60℃再搅拌2小时。冷却至室温后,将该反应混合物倒入饱和氯化铵中,并用EtOAc(2×500mL)提取。将组合的有机层用卤水洗涤,用硫酸钠干燥,过滤,浓缩,提供粗品化合物SB-E(7.36g,100%)。粗品不用进一步纯化,在下一步中使用。合成化合物SB-F.将粗品化合物SB-E(7.36g,21.9mmol)的THF(50mL)溶液用1NHCl水溶液酸化至pH=3。在室温搅拌12小时之后,将该反应混合物用乙酸乙酯(250mLx3)提取。用卤水洗涤组合的有机层,用硫酸钠干燥,过滤,并浓缩。用柱色谱纯化残余物(石油醚/乙酸乙酯=30:1至20:1),提供化合物SB-F(4.8g,16.7mmol,76%,两步)。1HNMR(400MHz,CDCl3)δ(ppm),5.12-5.10(m,1H),0.77(s,3H)。合成化合物SB-G.在0℃,通过注射泵,用30分钟向MeMgBr(28mmol,1M,在THF中)的THF(50mL)溶液中加入化合物SB-F(4.8g,16.8mmol)的干THF(10mL)溶液。在0℃搅拌5小时之后,将该反应混合物加热,并在室温搅拌过夜。将该反应混合物用冰冻水淬灭,并用乙酸乙酯(150mLx3)提取。用卤水洗涤组合的有机层,用硫酸钠干燥,过滤,并浓缩。用快速柱色谱纯化白色残余物(石油醚/乙酸乙酯=20:1至10:1),得到化合物SB-G(2.5g,8.28mmol,49%;Rf=0.35,石油醚/乙酸乙酯=10:1)。1HNMR(400MHz,CDCl3)δ(ppm),5.05-5.03(m,1H),1.21(s,3H),0.90(s,3H)。合成化合物SB-H.向化合物SB-G(2g,6.62mmol)的干THF(50mL)溶液中加入硼烷-四氢呋喃复合物(20mL;1.0M溶液,在THF中)。在室温搅拌1小时之后,将该反应混合物在冰浴中冷却,然后慢慢地用10%NaOH水溶液(10mL)、而后30%H2O2水溶液(12mL)猝灭。在室温搅拌一个小时之后,将该混合物用EtOAc(3×100mL)提取。将组合的有机层用10%Na2S2O3水溶液(100mL)、卤水(100mL)洗涤,用MgSO4干燥,过滤,并浓缩,提供粗品化合物SB-H(2g,100%)。粗品不用进一步纯化,在下一步中使用。合成化合物SB-I.向粗品化合物SB-H(2g,6.62mmol)的60mL湿润二氯甲烷(二氯甲烷与几毫升水一起摇动,然后与水层分离)溶液中加入Dess-Martin过碘化物(5.5g,13mmol)。在室温搅拌24小时之后,将该反应混合物用二氯甲烷(3×100mL)提取。将组合的有机层用10%Na2S2O3水溶液(100mL)、卤水(100mL)洗涤,用MgSO4干燥,过滤,并浓缩。用硅胶色谱纯化残余物(石油醚/乙酸乙酯=10:1至5:1),提供标题化合物SB-I(1g,3.14mmol,47%,两步)的灰白色固体。1HNMR(400MHz,CDCl3)δ(ppm),2.56(t,1H),2.11(s和m,4H),2.0(dt,1H),1.8(dm,2H),1.54(M,6H),1.43(m,1H),1.34(m,2H),1.20(m,12H),0.7(m,2H),0.62(s,3H)。合成化合物SB.向化合物SB-I(600mg,1.89mmol)的MeOH(20mL)溶液中加入5滴HBr(48%),而后加入溴(302mg,1.89mmol)。在室温搅拌1小时之后,将该反应混合物倒入冰-水中,然后用乙酸乙酯(100mLx3)提取。将组合的有机层用卤水(200mL)洗涤,用MgSO4干燥,过滤,浓缩,得到粗品化合物SB(600mg)。实施例32:合成化合物SB-1。向THF(5mL)中的K2CO3(25mg,0.18mmol)悬浮液添加1,2,4-三唑(13mg,0.18mmol)和化合物SB(36mg,0.09mmol)。在室温搅拌15h后,将反应混合物倒入5mLH2O中并且用EtOAc(2×10mL)提取。用卤水清洗组合的有机层,通过硫酸钠干燥,过滤并且浓缩。通过反相prep-HPLC纯化反应混合物以提供标题化合物,为灰白色固体(15mg,42%)。SB-1:1HNMR(500MHz,CDCl3),δ(ppm),8.14(s,1H),7.96(s,1H),5.02(AB,1H),4.93(AB,J=18.0Hz,1H),2.63(t,1H),1.21(s,CH3),0.69(s,3H)。实施例33:合成化合物SB-2。向THF(5mL)中的K2CO3(25mg,0.18mmol)悬浮液添加四唑(13mg,0.18mmol)和化合物SB(36mg,0.09mmol)。在室温搅拌15h后,将反应混合物倒入5mLH2O中并且用EtOAc(2×10mL)提取。用卤水清洗组合的有机层,通过硫酸钠干燥,过滤并且浓缩。通过反相prep-HPLC纯化反应混合物以提供SB-2,为灰白色固体(7mg,19%),和灰白色固体副产物(4mg,11%)。SB-2:1HNMR(500MHz,CDCl3),δ(ppm),8.58(s,1H),5.49(AB,1H),5.44(AB,1H),2.63(t,1H),1.21(s,CH3),0.72(s,3H)。实施例34:合成化合物SB-4和SB-5。向THF(5mL)中的K2CO3(67mg,0.50mmol)悬浮液添加5-甲基-1H-四唑(42.0mg,0.50mmol)和化合物SB(100mg,0.25mmol)。在室温搅拌15h后,将反应混合物倒入5mLH2O中并且用EtOAc(2×10mL)提取。用卤水(2×10mL)清洗组合的有机层,通过硫酸钠干燥,过滤并且在真空中浓缩。通过反相prep-HPLC纯化残留物以提供SB-4,为灰白色固体(10.1mg,0.025mmol,10.1%),和SB-5,为灰白色固体(21.3mg,0.053mmol,21.2%)。SB-4:1HNMR(500MHz,CDCl3)δ(ppm):5.12(AB,1H),5.06(AB,1H),2.66(t,1H),2.47(s,3H),1.21(s,CH3),0.69(s,3H).LCMS:Rt=2.19min.m/z=401.3[M+H]+。SB-5:1HNMR(500MHz,CDCl3)δ(ppm):5.35(AB,1H),5.34(AB,1H),2.63(t,1H),2.56(s,3H),1.21(s,CH3),0.72(s,3H).LCMS:Rt=2.30min.m/z=401.3[M+H]+。实施例35:合成化合物SB-6。向THF(5mL)中的K2CO3(25mg,0.18mmol)悬浮液添加1,2,3-1H-三唑(13mg,0.18mmol)和化合物SB(36mg,0.09mmol)。在室温搅拌15h后,将反应混合物倒入5mLH2O中并且用EtOAc(2×10mL)提取。用卤水清洗组合的有机层,通过硫酸钠干燥,过滤并且浓缩。通过反相prep-HPLC纯化反应混合物以提供SB-6,为灰白色固体(12mg,33%)。SB-6:1HNMR(500MHz,CDCl3),δ(ppm),7.76(s,1H),7.64(d,1H),5.26(AB,1H),5.14(AB,1H),2.59(t,1H),1.21(s,3H),0.68(s,3H)。实施例36:合成SD和SD中间体。合成化合物SD-B1和SD-B2。在-78℃在N2下向THF(25mL)和HMPA(0.5mL)中的化合物SC(1.3g,4.5mmol)和PhSO2CH2F(790mg,4.5mmol)的溶液逐滴添加LHMDS(5.5mL,1MinTHF)。在–78℃搅拌2h后,用饱和水性NH4Cl溶液(10mL)淬灭反应混合物并且允许加热到室温然后用Et2O(20mL×3)提取。用卤水清洗组合的有机层,通过硫酸钠干燥,过滤并且浓缩。通过硅胶柱层析纯化残留物(石油醚/乙酸乙酯=10/1)以给出化合物SD-B1和SD-B2(1.53g)的混合物。通过手性-HPLC进一步纯化混合物以提供化合物SD-B1-A(220mg,t=3.41min)。1HNMR(500MHz,CDCl3),δ(ppm),7.99-7.97(m,2H),7.75-7.74(m,1H),7.62-7.55(m,2H),5.13-5.09(m,1H),4.86-4.78(d,1H),0.88(s,3H);SD-B1-B(200mg,t=3.66min);1HNMR(500MHz,CDCl3),δ(ppm),7.96-7.95(m,1H),7.71-7.69(m,1H),7.62-7.58(m,2H),5.13-5.09(m,1H),4.87-4.77(d,1H),0.88(s,3H);SD-B2-A(235mg,t=4.9min).1HNMR(500MHz,CDCl3),δ(ppm),7.99-7.97(m,1H),7.72-7.70(m,1H),7.62-7.59(m,2H),5.29-5.20(d,1H),4.88-4.78(m,1H),0.88(s,3H);SD-B2-B(220mg,t=5.2min).1HNMR(500MHz,CDCl3),δ(ppm),7.99-7.97(m,2H),7.72(m,1H),7.62-7.59(m,2H),5.30-5.20(d,1H),5.09-5.08(m,1H),0.88(s,3H)。合成化合物SD-C。在-20℃在N2下向无水甲醇(15mL)中的化合物SD-B1-A(200mg,0.434mmol)和无水Na2HPO4(100mg)的溶液添加Na/Hg汞齐(400mg)。在–20℃至0℃搅拌1h后,弃去甲醇溶液,并且用Et2O(5x3mL)清洗固体残留物。在真空下除去组合有机相的溶剂,并且添加20ml卤水,接着用Et2O提取。用MgSO4干燥组合的醚相,并且除去醚以给出粗制产物,其通过硅胶层析(PE/EA=10/1)进一步纯化以给出产物99mg,69%。1HNMR(500MHz,CDCl3),δ(ppm),5.12-5.10(m,1H,),4.21-24.11(d,2H),0.88(s,3H).合成化合物SD-D。向干THF(5mL)中的化合物SD-C(95mg,0.296mmol)溶液添加硼烷-四氢呋喃复合物(1mL在THF中的1.0M溶液)。在室温搅拌1小时后,在冰浴中冷却反应混合物,然后用10%水性NaOH(1mL),接着用H2O2的30%水溶液(1.2mL)缓慢淬灭。允许混合物在室温搅拌1小时然后,用EtOAc(3x10mL)提取。用10%水性Na2S2O3(10mL),卤水(10mL)清洗组合的有机层,通过MgSO4干燥,过滤并浓缩以提供化合物SD-D(120mg粗制)。在没有进一步纯化的情况下将粗制产物用于下一步骤。合成化合物SD-E。向10mL湿二氯甲烷(二氯甲烷已与几毫升H2O一起摇动,然后与水层分离)中溶解的化合物SD-D(120mg粗制)溶液添加Dess-Martin过碘化物(300mg,707mmol)。在室温搅拌24h后,用二氯甲烷(3x10mL)提取反应混合物。用10%水性Na2S2O3(10mL),卤水(10mL)清洗组合的有机层,通过MgSO4干燥,过滤并且浓缩。通过硅胶上层析(石油醚/乙酸乙酯=1:5)纯化残留物以提供化合物SD-E(70mg,对于两步为70%),为灰白色固体。1HNMR(500MHz,CDCl3),δ(ppm),4.21-4.11(d,2H),2.19(s,3H),0.62(s,3H)。合成化合物SD。向甲醇(5mL)中的反应物(200mg,0.594mmol)溶液添加48%氢溴酸(300mg,1.782mmol),接着是溴(475mg,0.152mL,2.97mmol)。在25℃加热溶液达2小时。然后,将混合物倒入冷却水(50mL)。用乙酸乙酯(2×100mL)提取所得的固体。用卤水(100mL)清洗组合的有机提取物,通过硫酸镁干燥并且在真空中浓缩。在没有进一步纯化的情况下将粗制产物混合物直接用于下一步骤。实施例37:合成化合物SD-1。向THF(10mL)中的K2CO3(63mg,0.47mmol)悬浮液添加1,2,3-1H-三唑(11.4mg,0.47mmol)和化合物SD(100mg,0.23mmol)。在室温搅拌15h后,将反应混合物倒入5mLH2O中并且用EtOAc(2×10mL)提取。用卤水(2×10mL)清洗组合的有机层,通过硫酸钠干燥,过滤并在真空下浓缩。通过反相prep-HPLC纯化残留物以提供SD-1,为灰白色固体(28.7mg,29.5%),和SGE-00921-01-A,为灰白色固体(22.8mg,23.4%)。SD-1:1HNMR(500MHz,CDCl3),δ(ppm),7.76(d,1H),7.65(d,1H),5.28(AB,1H),5.14(AB,1H),4.17(d,2H),2.66(t,1H),0.68(s,3H).LCMS:Rt=2.18min.m/z=404.2[M+H]+。实施例38:合成化合物SD-2和SD-3。向THF(10mL)中的K2CO3(63mg,0.47mmol)悬浮液添加5-甲基-1H-四唑(39.5mg,0.47mmol)和化合物SD(100mg,0.24mmol)。在室温搅拌15h后,将反应混合物倒入5mLH2O中并且用EtOAc(2×10mL)提取。用卤水(2×10mL)清洗组合的有机层,通过硫酸钠干燥,过滤并且在真空中浓缩。通过反相prep-HPLC纯化残留物以提供SD-2,为灰白色固体(6.5mg,0.016mmol,6.7%),和SD-3,为灰白色固体(25.8mg,0.062mmol,25.8%)。SD-2:1HNMR(500MHz,CDCl3)δ(ppm):5.12(AB,1H),5.06(AB,1H),4.17(d,J=47.8Hz,2H),2.67(t,1H),2.47(s,3H),0.69(s,3H).LCMS:Rt=2.11min.m/z=419.3[M+H]+。SD-3:1HNMR(500MHz,CDCl3)δ(ppm):5.35(AB,1H),5.34(AB,1H),4.17(d,2H),2.63(t,1H),2.56(s,3H),0.72(s,3H).LCMS:Rt=2.21min.m/z=419.3[M+H]+。实施例39:合成化合物SD-4和SD-5。向无水THF(5mL)中的粗制反应物11(100mg,0.241mmol)溶液添加缺词?(140mg,1.2mmol),接着是碳酸钾(85mg,1.2mmol)。在60℃加热溶液2h,然后将溶液冷却到室温,并且用乙酸乙酯(100mL)稀释。用卤水(2×50mL)清洗所得的溶液,通过硫酸镁干燥并且在真空中浓缩。通过反相prep-HPLC纯化粗制产物以提供产物SD-4(15mg,0.04mmol,收率=17%)和灰白色固体副产物(26mg,0.06mmol,收率=25%)。SD-4:1HNMR(500MHz,CDCl3)δ(ppm):8.75(1H,s),5.32(1H,AB,J=18.5Hz),5.18(1H,AB),4.17(2H,d),2.68(1H,t),0.68(3H,s).LCMS:rt=2.14min,m/z=405[M+H]+。实施例40:合成SP和SP中间体。合成化合物SP-B。在0℃逐滴向干THF(50mL)中的反应物SC(4.4g,15.38mmol)溶液添加溴化乙基镁(ethylmagnesiumbromide)(THF中的3M,51.28mL)。然后,缓慢加热溶液,并且在周围温度搅拌15h。添加饱和NH4Cl溶液(20mL)以淬灭反应,并且用乙酸乙酯(3×100mL)提取所得的溶液。用卤水清洗提取物,通过Na2SO4干燥并且在真空中浓缩。通过快速层析(洗脱剂:石油醚:乙酸乙酯=10:1)纯化残留物以提供产物SP-B(3.15g,10.00mmol,64.8%),为灰白色固体。合成化合物SP-C。在室温向无水THF(10mL)中的反应物SP-B(500mg,1.58mmol)溶液添加BH3.THF(1.0M,7.23mL,7.23mmol),并且在25℃搅拌溶液过夜。然后,通过添加水(5mL),添加2MNaOH溶液(10mL),接着添加30%H2O2(10mL)淬灭反应。在室温将所得的混合物搅拌1小时。然后,用乙酸乙酯(200mL)稀释混合物,并且用卤水(2×100mL)清洗所得的溶液,通过硫酸镁干燥并且在真空中浓缩。在没有进一步纯化的情况下将粗制产物SP-C直接用于下一步骤。合成化合物SP-D。向冰水冷却浴中冷却的无水DCM(100mL)中的反应物SP-C(6.53g,19.67mmol)溶液按份添加氯铬酸吡啶(8.48g,39.34mol)。在周围温度将混合物搅拌过夜。然后,用DCM(50mL)稀释溶液并且过滤。用卤水(100mL)清洗组合的有机溶液,通过Na2SO4干燥并且在真空中浓缩。通过快速层析(洗脱剂:石油醚:乙酸乙酯=10:1)纯化残留物以提供产物SP-D(2.5g,7.53mmol,收率39%),为灰白色固体。SP-D:1HNMR(500MHz,CDCl3)δ(ppm):2.54(1H,t),2.11(3H,s),1.42-1.45(2H,q),0.91(3H,t),0.62(3H,s).合成化合物SP。向甲醇(5mL)中的反应物SP-D(80mg,0.24mmol)溶液添加48%氢溴酸(148mg,0.884mmol),接着是溴(241mg,0.077mL,1.505mmol)。在25℃将溶液加热1.5小时,然后,将混合物倒入冷却水(50mL)。用乙酸乙酯(2×50mL)提取所得的固体。用卤水(20mL)清洗组合的有机提取物,通过硫酸镁干燥并且在真空中浓缩。在没有进一步纯化的情况下将粗制产物SP直接用于下一步骤。实施例41:合成化合物SP-1和SP-2。向无水THF(10mL)中的粗制反应物SP(500mg,1.2mmol)溶液添加1,2,4-1H-三唑(500mg,6.0mmol),接着是碳酸钾(1.02g,6mmol)。在60℃加热溶液2h,然后将溶液冷却到室温,并且用乙酸乙酯(100mL)稀释。用卤水(2×50mL)清洗所得的溶液,通过硫酸镁干燥并且在真空中浓缩。通过反相prep-HPLC纯化粗制产物以提供产物SP-1(105mg,0.26mmol,收率=22%)和SP-2(62mg,0.15mmol,收率=13%),为灰白色固体。SP-1:1HNMR(500MHz,CDCl3)δ(ppm):7.75(1H,s),7.64(1H,s),5.26(1H,AB),5.14(1H,AB),2.66(1H,t),0.91(3H,t),0.68(3H,s).LCMS:rt=2.35min,m/z=400[M+H]+。SP-2:1HNMR(500MHz,CDCl3)δ(ppm):7.68(2H,s),5.25(1H,AB),5.23(1H,AB,2.59(1H,t),0.91(3H,t),0.70(3H,s).LCMS:rt=2.49min,m/z=400[M+H]+。实施例42:合成化合物SP-3。向THF(5mL)中的粗制反应物SP(247.5mg,0.603mmol,理论量)溶液添加四唑(84mg,1.202mmol),接着是碳酸钾(166mg,1.202mmol),并且在50℃将混合物加热2小时。然后,用乙酸乙酯(100mL)稀释反应混合物。用卤水(2×50mL)清洗所得的溶液,通过硫酸镁干燥并且在真空中浓缩。通过反相prep-HPLC纯化残留物以提供期望的产物SP-3(14.4mg,0.0359mmol,收率=6.0%(2步)),为灰白色固体。在prep-HPLC纯化中没有获得另一种期望的产物,由于其非常弱的吸收(214nm,254nm)所致。SP-3:1HNMR(400MHz,CDCl3)δ(ppm):8.57(1H,s),5.46(1H,AB),5.45(1H,AB),2.65(1H,t),1.45(2H,q),0.91(3H,t),0.73(3H,s).LCMS:rt=2.48min,m/z=401.1[M+H]+。实施例43:合成化合物SP-4和SP-5。向THF(5mL)中的K2CO3(67mg,0.50mmol)悬浮液添加5-甲基-1H-四唑(42.0mg,0.50mmol)和化合物SP(100mg,0.24mmol)。在室温搅拌15h后,将反应混合物倒入5mLH2O中并且用EtOAc(2×10mL)提取。用卤水清洗组合的有机层(2×10mL),通过硫酸钠干燥,过滤并且在真空中浓缩。通过反相prep-HPLC纯化残留物以提供SP-4,为灰白色固体(15.2mg,0.037mmol,15.2%),和SP-5,为灰白色固体(13.3mg,0.032mmol,13.3%)。SP-4:1HNMR(500MHz,CDCl3)δ(ppm):5.13(AB,1H),5.05(AB,1H),2.66(t,1H),2.48(s,3H),0.91(t,1H),0.69(s,3H).LCMS:Rt=2.30min.m/z=415.3[M+H]+。SP-5:1HNMR(400MHz,CDCl3)δ(ppm):5.36(AB,1H),5.35(AB,1H),2.63(t,1H),2.58(s,3H),0.91(t,1H),0.72(s,3H)。LCMS:Rt=2.38min.m/z=415.3[M+H]+。实施例44:合成SI和SI中间体。合成化合物SI-B。向干THF(20mL)中的化合物SI-A(5g,15mmol)溶液添加硼烷-四氢呋喃复合物(30mL在THF中的1.0M溶液),并且在周围温度将反应混合物搅拌1小时,然后缓慢添加10%水性NaOH(56mL)。在冰中冷却混合物,并且缓慢添加H2O2的30%水溶液(67mL)。将周围温度将混合物搅拌1小时,然后用EtOAc(3x100mL)提取。用10%水性Na2S2O3(100mL),卤水(100mL)清洗组合的EtOAc提取物,通过MgSO4干燥。过滤和除去溶剂给出粗制产物3.2g,用于下一步骤反应。合成化合物SI-C。向THF(40mL)中的化合物SI-B(3.2g,9mmol)溶液添加2MHCl(3mL)。将反应溶液在RT搅拌12h,然后在减压下除去溶剂。通过硅胶层析(洗脱剂:石油醚/乙酸乙酯=10:1至5:1)纯化粗制靶标化合物以给出2.2g产物,为灰白色固体,收率:81.40%。合成化合物SI-D。向100mLDMSO中的三甲基碘化锍(6.43g,31.5mmol)的搅拌溶液添加60wt%NaH(1.26g,31.5mmol)。在室温(15℃)搅拌1h后,逐滴添加20mLDMSO中的化合物I-C(2.2g,7.2mmol)的溶液。在2.5h后,将反应混合物倒入冰冷的水中,并且用醚(100mLx3)提取。然后,用卤水(100mLx3)清洗组合的醚层,干燥(MgSO4),过滤,并且浓缩以给出粗制产物1.6g,用于下一步反应。合成化合物SI-E。在60mLH2O饱和CH2Cl2中溶解化合物SI-D(1.6g,5mmol)。(使用分离漏斗,CH2Cl2已经与几毫升H2O一起摇动,并且与水层分开)。添加DMP(4.2g,10mmol),并且将所得的反应混合物剧烈搅拌24h。用DCM(100mL)稀释反应溶液,用10%水性Na2S2O3(100mL),卤水(100mL)清洗,通过MgSO4干燥,过滤,并且浓缩。通过硅胶上层析(洗脱剂:石油醚/乙酸乙酯=20:1至10:1)纯化残留物以提供标题化合物(1.2g,3.79mmol,75%),为灰白色固体。1HNMR(400MHz,CDCl3)δ(ppm):2.63(s,1H),2.59(s,1H),2.12(s,3H),0.63(s,3H)。合成化合物SI-F1和SI-F2。在干甲醇(250mL)中溶解SI-E(1.2g,3.8mmol),并且添加Na(262mg,11.4mmol)。将溶液回流16h。蒸发掉甲醇,并且在二氯甲烷中溶解残留物,并且用H2O(3x50mL)和卤水(100mL)清洗,通过MgSO4干燥,过滤,并且浓缩。通过硅胶层析(洗脱剂:石油醚/乙酸乙酯=10:1至5:1)纯化粗制靶标化合物以给出SI-F1(300mg,25%),SI-F2(300mg,25%),为灰白色固体。SI-F1:1HNMR(400MHz,CDCl3)δ(ppm):3.39(s,3H),3.19(s,2H),2.54(t,1H),2.11(s,3H),0.61(s,3H)。SI-F2:1HNMR(400MHz,CDCl3)δ(ppm):3.39(s,5H),3.37(s,2H),2.52(t,1H),2.11(s,3H),0.62(s,3H)。合成化合物SI。用2滴HBr(48%),接着用溴(6滴)处理MeOH中的SI-F1(50mg,0.14mmol)溶液。在rt将混合物搅拌1h,并且倒入冰水中。用EA(50mL)提取混合物,并且通过硫酸钠干燥。过滤实施例45:合成SI-1。向THF(5mL)中的粗制反应物(245.3mg,0.574mmol,理论量)的溶液添加四唑(201mg,2.87mmol),接着是碳酸钾(397mg,2.87mmol)。在60℃将混合物加热过夜。然后,用乙酸乙酯(100mL)稀释溶液。用卤水(2×50mL)清洗所得的溶液,通过硫酸镁干燥并且在真空中浓缩。通过反相prep-HPLC纯化残留物以提供级份1和级份2。级份2是纯产物SI-1(27.5mg,0.066mmol,两步总收率=11.5%),为灰白色固体。另外,通过硅胶层析(洗脱剂:石油醚/乙酸乙酯=1:4)纯化级份1以提供灰白色固体副产物(8.2mg,0.0197mmol,两步总收率=3.49%)。SI-1:1HNMR(500MHz,CDCl3)δ(ppm):8.75(1H,s),5.32(1H,AB),5.21(1H,AB),3.39(3H,s),3.19(2H,s),2.67(1H,t),0.68(3H,s).LC-MS:rt=2.19min,m/z=417.3[M+H]+。实施例46:合成化合物SI-2。向THF(50mL)中的K2CO3(248mg,1.8mmol)悬浮液添加1,2,3-1H-三唑(130mg,1.8mmol)和化合物SI(400mg,0.94mmol)。在室温搅拌15h后,将反应混合物倒入50mLH2O中,并且用EtOAc(2×100mL)提取。用卤水清洗组合的有机层,通过硫酸钠干燥,过滤并且浓缩。通过反相prep-HPLC纯化反应混合物以提供SI-2,为灰白色固体(80mg,20%)。SI-2:1HNMR(500MHz,CDCl3),δ(ppm),7.76(d,1H),7.64(d,1H),5.27(AB,1H),5.13(AB,1H),3.39(s,3H),3.19(s,2H),2.66(t,1H),0.68(s,3H)。实施例47:合成化合物SI-3和SI-4。向THF(5mL)中的K2CO3(67mg,0.50mmol)悬浮液添加5-甲基-1H-四唑(42.0mg,0.50mmol)和化合物SI(100mg,0.23mmol)。在室温搅拌15h后,将反应混合物倒入5mLH2O中并且用EtOAc(2×10mL)提取。用卤水(2×10mL)清洗组合的有机层,通过硫酸钠干燥,过滤并且在真空中浓缩。通过反相prep-HPLC纯化残留物以提供SI-3,为灰白色固体(12.6mg,0.029mmol,12.7%),和SI-4,为灰白色固体(22.3mg,0.052mmol,22.5%)。SI-3:1HNMR(500MHz,CDCl3)δ(ppm):5.13(AB,1H),5.05(AB,1H),3.39(s,3H),3.19(s,2H),2.66(t,1H),2.47(s,3H),0.69(s,3H).LC-MS:Rt=2.14min.m/z=431.3[M+H]+。SI-4:1HNMR(500MHz,CDCl3)δ(ppm):5.35(AB,1H),5.34(AB,1H),3.39(s,3H),3.19(s,2H),2.63(t,1H),2.56(s,3H),0.72(s,3H).LC-MS:Rt=2.25min.m/z=401.3[M+H]+。实施例48:合成SQ和SQ中间体。合成化合物SQ-B。向100mLDMSO中的三甲基碘化锍(8.1g,36.9mmol)的搅拌溶液添加NaH(60%;1.26g,31.5mmol)。在室温搅拌1h后,逐滴添加DMSO(20mL)中的化合物C(2.2g,7.2mmol)的悬浮液。将混合物再搅拌2.5h,然后倒入冰冷的水中,并且用醚(100mLx3)提取。然后用卤水(100mLx3)清洗组合的醚层,通过MgSO4干燥,过滤,并且浓缩以给出粗制产物SQ-B(2.2g)。在没有进一步纯化的情况下将粗制产物用于下一步骤。合成化合物SQ-C。在干甲醇(250mL)中溶解化合物SQ-B(2.2g,7.3mmol),并且添加Na(672mg,29.2mmol)。将溶液搅拌回流6h。蒸发掉甲醇,并且在二氯甲烷中溶解残留物,并且用H2O(3x50mL)和卤水(100mL)清洗,通过MgSO4干燥,过滤,并且浓缩。通过硅胶层析(石油醚/乙酸乙酯=10:1至5:1)纯化粗制靶标化合物,并且浓缩以给出SQ-C(1.8g,82%),为灰白色固体。1HNMR(500MHz,CDCl3),δ(ppm),5.03-5.01(m,1H),3.43(q,2H),3.13(s,2H),0.80(s,3H)。合成化合物SQ-D。向干THF(50mL)中的化合物SQ-C(1.8g,5.2mmol)溶液添加硼烷-四氢呋喃复合物(20mL在THF中的1.0M溶液)。在室温搅拌1小时后,在冰浴中冷却反应混合物,然后用10%水性NaOH(10mL),接着用H2O2的30%水溶液(12mL)缓慢淬灭。允许混合物在室温搅拌1小时,然后用EtOAc(3x100mL)提取。用10%水性Na2S2O3(100mL),卤水(100mL)清洗组合的有机层,通过MgSO4干燥,过滤并浓缩以提供粗制化合物SQ-D(1.8g,100%)。在没有进一步纯化的情况下将粗制产物用于下一步骤。合成化合物SQ-E。向60mLH2O饱和二氯甲烷(二氯甲烷已经与几毫升H2O一起摇动,然后与水层分离)中溶解的粗制化合物SQ-D(1.8g,5.2mmol)溶液添加Dess-Martin过碘化物(4.4g,10.4mmol)。在室温搅拌24h后,用二氯甲烷(3x100mL)提取反应混合物。用10%水性Na2S2O3(100mL),卤水(100mL)清洗组合的有机层,通过MgSO4干燥,过滤并且浓缩。通过硅胶上层析(石油醚/乙酸乙酯=10:1至5:1)纯化残留物以提供SQ-E(1g,2.8mmol,对于两步为56%),为灰白色固体。1HNMR(400MHz,CDCl3),δ(ppm),3.52(q,2H),3.21(s,2H),2.54(t,2H),2.11(s,3H),1.20(t,3H),0.61(s,3H).LCMS:Rt=7.25min.m/z=345.1[M-17]+。合成化合物SQ。向MeOH(20mL)中的化合物SQ-E(600mg,1.65mmol)溶液添加5滴HBr(48%),接着是溴(264mg,1.65mmol)。在室温搅拌1h后,将反应混合物倒入冰水中,然后用乙酸乙酯(100mLx3)提取。用卤水(200mL)清洗组合的有机层,通过MgSO4干燥,过滤并浓缩以给出粗制化合物SQ(600mg,100%)。在没有进一步纯化的情况下将粗制产物用于下一步骤。LCMS:Rt=7.25min.m/z=463.1[M+Na]+。实施例49:合成化合物SQ-1和SQ-2。向THF(10mL)中的K2CO3(188mg,1.36mmol)悬浮液添加1,2,3-1H-三唑(94mg,1.36mmol)和化合物SQ(300mg,0.68mmol)。在室温搅拌15h后,将反应混合物倒入5mLH2O中并且用EtOAc(2×10mL)提取。用卤水清洗组合的有机层(2×10mL),通过硫酸钠干燥,过滤并在真空下浓缩。通过反相prep-HPLC纯化残留物以提供SQ-1,为灰白色固体(81mg,0.19mmol,27.9%),和SQ-2,为灰白色固体(41mg,0.10mmol,14.7%)。SQ-1:1HNMR(400MHz,CDCl3)δ(ppm):7.76(s,1H),7.64(s,1H),5.28(AB,1H),5.14(AB,1H),3.53(q,2H),3.22(s,2H),2.66(t,1H),1.20(t,3H),0.68(s,3H).LCMS:Rt=2.21min.m/z=430.3[M+H]+。SQ-2:1HNMR(400MHz,CDCl3)δ(ppm):7.69(s,2H),5.27(AB,1H),5.22(AB,1H),3.53(q,2H),3.22(s,2H),2.60(t,1H),1.20(t,3H),0.71(s,3H).LCMS:Rt=2.34min.m/z=430.3[M+H]+。实施例50:合成化合物SQ-3和SQ-4。向THF(10mL)中的K2CO3(94mg,0.68mmol)悬浮液添加1,2,3-1H-三唑(48mg,0.68mmol)和化合物SQ(150mg,0.34mmol)。在室温搅拌15h后,将反应混合物倒入5mLH2O中,并且用EtOAc(2×10mL)提取。用卤水清洗组合的有机层(2×10mL),通过硫酸钠干燥,过滤并在真空下浓缩。通过反相prep-HPLC纯化残留物以提供SQ-3,为灰白色固体(20.9mg,0.049mmol,14.4%)和SQ-4,为灰白色固体(15.2mg,0.035mmol,10.3%)。SQ-3:1HNMR(400MHz,CDCl3)δ(ppm):8.57(s,1H),5.46(AB,1H),5.45(AB,1H),3.53(q,2H),3.22(s,2H),2.66(t,1H),1.21(t,3H),0.72(s,3H).LCMS:Rt=2.35min.m/z=431.4[M+H]+。SQ-4:1HNMR(400MHz,CDCl3)δ(ppm):8.74(s,1H),5.32(AB,J=18.0Hz,1H),5.18(AB,J=18.1Hz,1H),3.52(q,2H),3.22(s,2H),2.68(t,1H),1.20(t,3H),0.68(s,3H).LCMS:Rt=2.22min.m/z=431.4[M+H]+。实施例51:合成化合物SQ-5和SQ-6。向THF(5mL)中的K2CO3(67mg,0.50mmol)悬浮液添加5-甲基-1H-四唑(42.0mg,0.50mmol)和化合物SQ(100mg,0.25mmol)。在室温搅拌15h后,将反应混合物倒入5mLH2O中并且用EtOAc(2×10mL)提取。用卤水(2×10mL)清洗组合的有机层,通过硫酸钠干燥,过滤并且在真空中浓缩。通过反相prep-HPLC纯化残留物以提供SQ-5,为灰白色固体(8.5mg,0.019mmol,8.1%)和SQ-6,为灰白色固体(14.8mg,0.034mmol,13.2%)。SQ-5:1HNMR(500MHz,CDCl3)δ(ppm):5.13(AB,1H),5.06(AB,1H),3.53(q,2H),3.22(s,2H),2.67(t,1H),1.21(t,3H),0.69(s,3H).LCMS:Rt=2.26min.m/z=445.4[M+H]+。SQ-6:1HNMR(500MHz,CDCl3)δ(ppm):5.36(AB,1H),5.35(AB,1H),3.53(q,2H),3.22(s,2H),2.64(t,1H),2.56(s,2H),1.20(t,3H),0.72(s,3H)。LCMS:Rt=2.35min.m/z=445.3[M+H]+。实施例52:合成SV和SV中间体。合成化合物SV-B。向600mLCH3CN中的SL-B(68g,216.27mmol)溶液,在-4℃按份添加selectflour(90.22g,324.4mmol)。在-4℃将所得反应混合物搅拌3h。在TLC显示完成反应后,然后将混合物过滤并且浓缩。通过硅胶上的柱层析纯化产物,用(石油醚/乙酸乙酯20:1-15:1-10:1-8:1-6:1-5:1)洗脱,以提供SV-B(26.3g,41.8%收率),为灰白色固体。1HNMR(SV-B)(400MHz,CDCl3),δ(ppm),6.02-5.94(m,1H,),5.20-5.01(m,1H),2.55-2.26(m,6H),2.16-2.05(m,1H),2.01-1.83(m,4H),1.48-1.22(m,5H),0.98-0.78(m,6H)。合成化合物SB-X。在20℃向EtOAc(350mL)中的SV-B(27g,92.98mmol)溶液,然后在混合物中添加Pd/C(2.7g,5%)。在氢气下在20℃,1atm将溶液搅拌10h。在LCMS显示完成反应后,然后将混合物过滤并且浓缩。通过在硅胶上的柱层析纯化产物,用(石油醚/乙酸乙酯40:1-35:1-30:1-25:1-20:1-15:1-10:1-6:1)洗脱以给出SB-X(15.6g,56.38%),为灰白色固体。1HNMR(SB-X)(400MHz,CDCl3),δ(ppm)=4.68-4.56(m,1H),2.64-2.51(m,1H),2.53-2.03(m,8H),1.97-1.80(m,4H),1.49-1.20(m,6H),0.96-0.92(m,2H),0.88-0.78(m,1H)。合成化合物SB-Y。在23℃向MeOH(600mL)中的SB-X(47g,160.75mmol)溶液,然后在混合物中添加2.35gTsOH。在60℃将溶液搅拌1.5h。在TLC显示完成反应后,然后将混合物过滤并浓缩以给出SB-Y(35g,64.33%),为灰白色固体。1HNMR(SB-Y)(400MHz,CDCl3),δ(ppm)=4.74-4.57(m,1H),3.16(s,3H),3.10(s,3H),2.47-2.35(m,1H),2.15-2.09(m,1H),2.06-1.82(m,6H),1.77-1.15(m,11H),1.05-0.96(m,1H),0.89(s,3H),0.83-0.77(m,1H)。合成化合物SB-Z。向150mLTHF中的乙基三苯基溴化膦(115.17g,310.23mmol)溶液,添加KOt-Bu(34.81g,310.23mmol)。将反应混合物加热到60℃达1h,并且将SB-Y(35g,103.41mmol)添加到混合物,将该混合物于60℃再搅拌15h。冷却反应混合物,并且用1500mLEtOAc提取,用卤水清洗,并且浓缩以提供SB-Z,为灰白色固体(120g,粗制)。1HNMR(SB-Z)(400MHz,CDCl3),δ(ppm)=5.13-5.07(m,1H),4.67-4.54(m,1H),3.14(s,3H),3.09(s,3H),2.42-2.15(m,3H),1.92-1.79(m,3H),1.67-1.61(m,4H),1.57-1.50(m,2H),1.45-1.15(m,10H),1.01-0.94(m,1H),0.92(s,3H),0.90-0.84(m,1H)。合成化合物SB-AA。向600mLTHF中的SB-Z(120g,粗制)溶液,添加2M含水HCl90mL。在22℃将反应混合物搅拌1h。在TLC显示完成反应后,然后用含水NaHCO3淬灭反应。用500mLEtOAc提取反应,用卤水清洗并且在真空中蒸发。通过层析(石油醚/乙酸乙酯=150:1-125:1-100:1-80:1-60:1-50:1)纯化所得的残留物以提供SB-AA,为灰白色固体(24g,76.23%收率)。1HNMR(SB-AA)(400MHz,CDCl3),δ(ppm)=5.13(m,1H),4.65-4.48(m,1H),2.62-2.42(m,1H),2.44-2.07(m,8H),1.92-1.80(m,1H),1.72-1.55(m,8H),1.36-1.08(m,6H),0.92(s,3H),0.83-0.73(m,1H)。合成化合物SB-BB。向50mLTHF中的Me3SOI(78.07g,354.75mmol)溶液,添加50mLTHF中的t-BuOK(39.81g,354.75mmol)溶液。在60℃将反应混合物搅拌1.5h。然后,在反应中添加THF(300mL)中的SB-AA(24g,78.83mmol)溶液。在23℃将反应搅拌2.5h。在TLC显示完成反应后,然后用冰水淬灭反应。用500mLEtOAc提取反应,用卤水清洗并且在真空中蒸发以提供SB-BB,为粗制产物(50g)。1HNMR(SB-BB)(400MHz,CDCl3),δ(ppm)=5.20-5.11(m,1H),4.65-4.52(m,1H),2.74-2.68(m,2H),2.48-1.81(m,9H),1.72-1.64(m,4H),1.55-1.06(m,10H),0.97-0.89(m,3H),0.85-0.77(m,1H)。合成化合物SB-CC。向300mLTHF中的SB-BB(50g,粗制)溶液,在0℃添加LiAlH4(8.99g,236.49mmol)。在23℃将反应混合物搅拌1.5h。在TLC显示完成反应后,然后用水淬灭反应。用1000mLEtOAc提取反应,用卤水清洗并且在真空中蒸发。通过层析(石油醚/乙酸乙酯=100:1-80:1-60:1-50:1-40:1-30:1)纯化所得的残留物以提供SB-CC,为灰白色固体(19g,75.19%收率)。1HNMR(SB-CC)(400MHz,CDCl3),δ(ppm)=5.17-5.07(m,1H),4.66-4.48(m,1H),2.41-2.32(m,1H),2.28-2.15(m,2H),2.09-2.05(m,1H),1.88-1.75(m,2H),1.68-1.64(m,3H),1.40-1.31(m,1H),1.25-1.13(m,9H),0.89(s,3H),0.81-0.72(m,1H)。合成化合物SB-DD。在0℃向干THF(500mL)中的SB-CC(19g,59.29mmol)溶液添加C2H9BS(59.29mL;THF中的10M溶液)。在室温搅拌2小时后,在冰浴中冷却反应混合物,然后用3M水性NaOH(160mL),接着用H2O2的30%水溶液(100mL)缓慢淬灭。在20℃搅拌1.5h后,过滤混合物,并且用EtOAc(300mL)提取。用含水Na2S2O3处理组合的有机层,提取,干燥,并且浓缩以提供SB-DD,为粗制(21g,粗制)。在没有进一步纯化的情况下将粗制产物用于下一步骤。合成化合物SB-EE。向200mLCH2Cl2中的SB-DD(21g,59.29mmol)溶液,在0℃添加PCC(25.56g,118.58mmol),在22℃搅拌2h。过滤反应混合物,并且用20mLCH2Cl2提取,用含水NaHCO3,含水Na2S2O3,卤水清洗,并且在真空中蒸发。通过层析(石油醚/乙酸乙酯=15:1-10:1-6:1)纯化残留物以提供SB-EE,为灰白色固体(12g,60.15%收率)。1HNMR(SB-EE)(400MHz,CDCl3),δ(ppm)=4.65-4.46(m,1H),2.55-2.51(m,1H),2.22-2.09(m,4H),2.06-1.97(m,32H),1.88-1.77(m,2H),1.69-1.54(m,5H),1.48-1.30(m,3H),1.28-1.05(m,11H),0.83-0.72(m,1H),0.63(s,3H)。合成化合物SV。向1500mLMeOH中的SB-EE(12g,35.66mmol)溶液,在0℃添加HBr(5滴)和Br2(2.01mL,39.23mmol)。在16℃将反应搅拌2h。用含水NaHCO3淬灭反应混合物,并且浓缩。然后,用1000mlEtOAc提取混合物,用卤水清洗并且在真空中蒸发。通过在硅胶上的柱层析纯化产物,用(石油醚/乙酸乙酯=12:1-10:1-8:1-6:1-3:1)洗脱,以提供SV,为灰白色固体(12.3g,83.03%收率)。1HNMR(SV)(400MHz,CDCl3),δ(ppm)=4.64-4.47(m,1H),3.95-3.86(m,2H),2.89-2.80(m,1H),2.23-2.16(m,1H),2.07-1.64(m,8H)1.46-1.06(m,14H),0.83-0.74(m,1H),0.67(s,3H)。实施例53:合成化合物SV-1和SV-2。向THF(5mL)中的SV(40mg,0.09mmol)的悬浮液添加1H-1,2,3-三唑(30mg,0.45mmol)和K2CO3(60mg,0.45mmol)。在25℃将混合物搅拌15h。然后,用乙酸乙酯(100mL)稀释溶液,并且用卤水(100mL)清洗所得的溶液,通过硫酸钠干燥并且在真空中浓缩。通过反相prep-HPLC纯化反应混合物以提供SV-1,为灰白色固体(10mg,26%收率),和SV-2,为灰白色固体(10mg,26%收率)。SV-1:1HNMR(400MHz,CDCl3),δ(ppm),7.75(s,1H),7.65(s,1H),5.29-5.25(1H,AB),5.25-5.17(1H,AB),4.61-4.52(d,1H),2.6(1H,t),1.18(s,3H),0.63(s,3H)。SV-2:1HNMR(400MHz,CDCl3),δ(ppm),7.68(s,2H),5.24-5.23(m,2H),4.60-4.50(d,1H),2.6(1H,t,),1.25(s,3H),0.74(s,3H)。实施例54:合成化合物SV-3和SV-4。向3mLDMF中的SV(100mg,0.24mmol)溶液添加2H-四唑(33.73mg,0.48mmol)和K2CO3(99.82mg,0.72mmol)。在28℃将反应搅拌2h。用水淬灭所得的溶液,并且用EtOAc(50mL)提取。用卤水(20mL)清洗有机层,通过Na2SO4干燥,并且在真空中浓缩。通过硅胶上的柱层析纯化残留物,用(PE/EA=12/1至2/1)洗脱,以给出SV-3(16.1mg,收率:16.67%)和SV-4(28.3mg,收率:29.17%),为灰白色固体。1HNMR(SV-3):(400MHz,CDCl3)δ8.60(s,1H),5.57-5.42(m,2H),4.73-4.48(m,1H),2.74-2.60(m,1H),2.31-2.21(m,1H),2.16-2.108(m,1H),1.97-1.89(m,1H),1.86-1.60(m,7H),1.55-1.11(m,14H),0.88-0.80(m,1H),0.77(s,3H).1HNMR(SV-4):(400MHz,CDCl3)δ8.75(s,1H),5.36-5.16(m,2H),4.66-4.47(m,1H),2.73-2.62(m,1H),2.30-2.18(m,1H),2.09-1.74(m,6H),1.67-1.60(m,3H),1.38-1.16(m,11H),0.88-0.75(m,1H),0.70(s,3H)。实施例55:合成化合物SV-5和SV-6。向3mLDMF中的SV(100mg,0.24mmol)溶液添加5-甲基-2H-四唑(40.48mg,0.48mmol)和K2CO3(99.82mg,0.72mmol)。在21℃将反应搅拌1h。用水淬灭所得的溶液,并且用EtOAc(50mL)提取。在真空中浓缩有机层。通过硅胶上的柱层析(PE/EtOAc=8/1至1/1)纯化残留物以给出SV-5(21.3mg,收率:21.14%)和SV-6(27.1mg,收率:26.89%),为灰白色固体。1HNMR(SV-5):(400MHz,CDCl3)δ5.43-5.31(m,2H),4.68-4.49(m,1H),2.69-2.62(m,1H),2.59(s,3H),2.31-2.20(m,1H),2.14-2.09(m,1H),1.95-1.88(m,1H),1.85-1.60(m,8H),1.46-1.20(m,12H),1.02-0.93(m,1H),0.89-0.80(m,1H),0.77(s,3H).1HNMR(SV-6):(400MHz,CDCl3)δ5.21-5.05(m,2H),4.69-4.50(m,1H),2.73-2.63(m,1H),2.50(s,3H),2.30-2.19(m,1H),2.13-2.01(m,2H),1.98-1.57(m,9H),1.45-1.14(m,12H),0.90-0.80(m,1H),0.73(s,3H)。实施例56:合成化合物SV-7。向15mLDMF中的SV(100mg,0.24mmol)溶液添加4-甲基-2H-1,2,3-三唑(40.01mg,0.48mmol)和K2CO3(99.82mg,0.72mmol)。在28℃将反应搅拌2h。用水淬灭所得的溶液并且用EtOAc(50mL)提取。用卤水(20mL)清洗有机层,通过Na2SO4干燥,并且在真空中浓缩。通过prep-HPLC纯化残留物以给出SV-7(20.6mg,收率:20.83%),为灰白色固体。1HNMR(SV-7):(400MHz,CDCl3)δ7.45(s,1H),5.23-5.10(m,2H),4.68-4.49(m,1H),2.64-2.57(m,1H),2.35(s,3H),2.30-2.18(m,1H),2.14-2.00(m,2H),1.93-1.58(m,8H),1.46-1.09(m,13H),0.86-0.76(m,1H),0.75(s,3H)。实施例57:合成化合物SV-8和SV-9。向10mLDMF(5mL)中的SV(200mg,0.48mmol)溶液添加4-甲基-2H-1,2,3-三唑(80.02mg,0.96mmol)和K2CO3(199.63mg,1.44mmol)。在17℃将反应混合物搅拌2h。用水淬灭所得的溶液并且用EtOAc(50mL)提取。干燥有机层,并且浓缩。通过硅胶纯化残留物以给出90mgSV-8/SV-9混合物和副产物(60mg)。通过SFC纯化分开混合物以给出SV-8(38.8mg,收率:29.84%)和SV-9(31.5mg,收率:23.3%),为灰白色固体。1HNMR(SV-8):(400MHz,CDCl3)δ7.347(s,1H),5.191-5.041(q,J1=17.6HMz,J2=42.4HMz),4.62-4.50(m,1H),2.66-2.61(m,1H),2.37(s,3H),2.10-2.06(m,1H),1.87-1.74(m,2H),1.70-1.50(m,7H),1.30-1.04(m,14H),0.86-0.76(m,1H),0.70(s,3H).1HNMR(SV-9):(400MHz,CDCl3)δ7.488(s,1H),5.08-5.07(m,2H),4.63-4.50(m,1H),2.68-2.63(m,1H),2.22(s,3H),2.04-1.89(m,2H),1.80-1.73(m,7H),1.64-1.60(m,1H),1.56-1.20(m,14H),0.80-0.70(m,1H),0.64(s,3H)。实施例58:合成SW和SW中间体。合成化合物SW-B。将SW-A(10g,36.7mmol)添加到50mL氯化乙酰和50mL乙酸酐。将反应混合物加热到120℃达5h,在真空中蒸发以提供SW-B,为灰白色固体(10g,87%收率)。1HNMR(400MHz,CDCl3),δ(ppm),5.78(s,1H),5.55(s,1H),2.4(dd,2H),2.13(s,3H),0.90(s,3H)。合成化合物SW-C。在0℃向200mLTHF和20mLH2O中的SW-B(10g,31.8mmol)溶液,添加mCPBA(11g,63.6mmol),在rt搅拌15h,用500mLEtOAc提取反应混合物,用100mL饱和Na2SO3,100ml饱和NaHCO3和100mL卤水清洗,并且在真空中蒸发,然后通过层析(PE:EtOAc=5:1)纯化以提供SW-C,为灰白色固体(2.2g,24%收率)。1HNMR(400MHz,CDCl3),δ(ppm),5.92(s,1H),4.44(s,1H),0.95(s,3H)。合成化合物SW-D。向50mLEtOAc中的SW-C(2g,6.94mmol)溶液,添加Pd/C200mg。在1atmH2将反应混合物氢化15h。然后,在真空中蒸发反应混合物,并且通过层析纯化(PE:EtOAc=1:2)以提供SW-D,为灰白色固体(0.5g,25%收率)。1HNMR(400MHz,CDCl3),δ(ppm),3.84(s,1H),2.62(1H,t)0.95(s,3H)。合成化合物SW-E。向100mLMeOH中的SW-D(1g,3.4mmol)溶液,添加TsOH50mg,加热到60℃达2h。用500mLEtOAc提取反应混合物,用100mL饱和NaHCO3,100mL卤水清洗,并且在真空中蒸发以提供SW-E,为灰白色固体(1g,91%收率)。合成化合物SW-F。向30mLTHF中的乙基三苯基溴化膦(10.67g,28.84mmol)溶液,添加KOt-Bu(3.23g,28.80mmol)。将反应加热到60℃达1h。添加SW-E(3.23g,9.6mmol),并且将所得的混合物在60℃搅拌15h。然后,用500mLEtOAc提取反应混合物,用卤水清洗,并且在真空中蒸发。通过层析(PE:EtOAc=3:1)纯化所得的粗制残留物以提供SW-F,为灰白色固体(2.17g,64%收率)。合成化合物SW-G。向50mLTHF中的SW-F(1g,2.9mmol)溶液,添加NaH(2g,5.8mmol),并且将所得的混合物在rt搅拌1h。然后,将1mLMeI添加到混合物,然后在rt搅拌过夜。用5mLH2O淬灭反应混合物,并且用100mLEtOAc提取,用卤水清洗并且在真空中蒸发。通过层析(PE:EtOAc=10:1)纯化所得的残留物以提供SW-G,为灰白色固体(587mg,59%收率)。合成化合物SW-H。向20mLTHF中的SW-G(1g,2.8mmol)溶液,添加2M含水HCl(2mL),并且将所得反应混合物在rt搅拌1h。然后用5mLH2O淬灭反应混合物,并且用100mLEtOAc提取,用卤水清洗并且在真空中蒸发。通过层析(PE:EtOAc=10:1)纯化残留物以提供SW-H,为灰白色固体(745mg,81%收率)。1HNMR(400MHz,CDCl3),δ(ppm),5.05-5.03(m,1H),3.24(s,3H),3.11(s,1H),2.6(1H,t),0.87(s,3H)。合成化合物SW-I。向5mLTHF中的三甲基碘化亚砜(3.6g,16.5mmol)的搅拌溶液添加叔丁醇钾(potassiumtert-butanolate)(1.90g,16.5mmol)。在60℃搅拌1.5h后,逐滴添加10mLTHF中的SW-H(1g,3.3mmol)的悬浮液。再过3h后,将反应混合物倒入冰冷的水中,并且用EtOAc(100mLx3)提取,用卤水(100mLx3)清洗,干燥(MgSO4),过滤,并且在真空中蒸发以提供SW-I,为灰白色固体(800mg,73%收率)。在没有进一步纯化的情况下将粗制产物用于下一步骤。合成化合物SW-J。向10mLTHF中的SW-I(150mg,0.45mmol)溶液,添加LiAH4(50mg,1.35mmol),将所得的反应混合物在rt搅拌1h。然后,用5mLH2O淬灭反应混合物,并且用100mLEtOAc提取,用卤水清洗并且在真空中蒸发。通过层析(PE:EA=3:1)纯化残留物以提供SW-J,为灰白色固体(108mg,72%收率)。1HNMR(400MHz,CDCl3),δ(ppm),5.12-5.10(m,1H),3.29(s,3H),3.18(s,1H),1.23(s,3H),0.88(s,3H)。合成化合物SW-K。向干THF(5mL)中的SW-J(100mg,0.3mmol)溶液添加硼烷-四氢呋喃复合物(1mL;THF中的1.0M溶液)。在室温搅拌1小时后,在冰浴中冷却反应混合物,然后用10%水性NaOH(1mL),接着用H2O2的30%水溶液(1mL)缓慢淬灭。在室温搅拌1小时后,用EtOAc(3x100mL)提取混合物。用10%水性Na2S2O3(100mL),卤水(100mL)清洗组合的有机层,通过MgSO4干燥,过滤并浓缩以提供SW-K,为灰白色固体(90mg,81%)。在没有进一步纯化的情况下将粗制产物用于下一步骤。合成化合物SW-L。向20mLDCM中的SW-K(100mg,0.29mmol)溶液,添加PCC(190mg,0.87mmol),在rt搅拌2h。用5mlH2O淬灭反应混合物,并且用100mLEtOAc提取,用卤水清洗并且在真空中蒸发,然后通过层析(PE:EtOAc=3:1)纯化以提供SW-L,为灰白色固体(52mg,51%收率)。1HNMR(400MHz,CDCl3),δ(ppm),3.26(s,3H),3.16(s,1H),2.11(s,3H),1.20(s,3H),0.61(s,3H)。合成化合物SW。向MeOH(5mL)中的SW-L(40mg,0.11mmol)溶液添加2滴HBr(48%),接着是溴(150mg,0.33mmol)。在室温搅拌1小时后,将反应混合物倒入冰水中,然后用乙酸乙酯(10mLx3)提取。用卤水(20mL)清洗组合的有机层,通过MgSO4干燥,过滤并浓缩以给出粗制化合物SW,为灰白色固体(40mg,80%收率)。在没有进一步纯化的情况下将粗制产物用于下一步骤。实施例59:合成化合物SW-1和SW-2。向THF(5mL)中的SW(40mg,0.09mmol)悬浮液添加1H-1,2,3-三唑(30mg,0.45mmol)和K2CO3(60mg,0.45mmol)。在25℃将混合物搅拌15h。然后,用乙酸乙酯(100mL)稀释溶液,并且用卤水(100mL)清洗所得的溶液,通过硫酸钠干燥并且在真空中浓缩。通过反相prep-HPLC纯化反应混合物以提供SW-1,为灰白色固体(10mg,26%收率),和SW-2,为灰白色固体(8mg,20%收率)。SW-1:1HNMR(400MHz,CDCl3),δ(ppm),7.75(s,1H),7.64(s,1H),5.27-5.24(1H,AB),5.17-5.13(1H,AB),3.28(s,3H),3.17(s,1H),2.7(1H,t),1.23(s,3H),0.65(s,3H).SW-2:1HNMR(400MHz,CDCl3),δ(ppm),7.68(s,2H),5.28-5.25(1H,AB),5.23-5.20(1H,AB),3.28(s,3H),3.17(s,1H),2.6(1H,t),1.24(s,3H),0.75(s,3H)。实施例60:合成SZ和SZ中间体。合成化合物SZ-B。在-78℃向THF(18mL)中的化合物SZ-A(500mg,1.82mmol)溶液添加LiHMDS(1.0MinTHF溶液,4.00mL,4.00mmol)。在-78℃将溶液搅拌30分钟。然后,添加HMPA(0.69mL,4.00mmol)。在-78℃将溶液再搅拌30分钟。然后,添加碘甲烷(0.34mL,5.46mmol)。在-78℃将溶液再搅拌2小时,并且加热到室温,并且搅拌1小时。通过添加水(2mL)淬灭反应。在真空中除去大部分THF溶剂。然后,用乙酸乙酯(100mL)稀释残留物,并且用卤水(2×100mL)清洗所得的溶液,通过硫酸镁干燥。在真空中除去溶剂提供粗制产物SZ-B(350mg,67%),为稠油(thickoil)。在没有进一步纯化的情况下将粗制产物用于下一步骤。SZ-B:1HNMR(500MHz,CDCl3)δ(ppm):5.74(1H,s),3.67(1H,t),1.11(3H,d),0.81(3H,s)。合成化合物SZ-C。在-78℃向液氨(100mL)添加锂(687mg,99.0mmol)。将液体转变为深蓝色。然后,将t-BuOH(244mg,3.30mmol)和THF(20mL)中的反应物SZ-B(950mg,3.30mmol)的溶液添加到Li-氨溶液。在-78℃将混合物搅拌4小时。然后,添加NH4Cl固体(7g)以淬灭反应。将混合物从深蓝色转变为白色。让混合物加热到室温,并且在通风柜中蒸发氨过夜。向残留物添加水(100mL)。通过浓HCl将混合物酸化为pH6-7。然后,添加乙酸乙酯(100mL)。用乙酸乙酯(2×100mL)进一步提取分离的水层。用卤水(200mL)清洗组合的有机提取物,通过硫酸镁干燥并且在真空中浓缩。在没有进一步纯化的情况下将粗制产物SZ-C直接用于下一步骤。合成化合物SZ-D。向二氯甲烷(60mL)中的粗制化合物SZ-C(980mg,3.40mmol)溶液添加重铬酸吡啶(重铬酸吡啶)(PDC)(2.56g,6.80mmol)。在室温将混合物搅拌过夜。将溶液过滤通过硅藻土(celite)的短垫。用CH2Cl2(3×50mL)清洗硅藻土。在真空中浓缩组合的CH2Cl2溶液。通过快速层析纯化残留物(洗脱剂:石油醚/EtOAc=5:1)以提供产物SZ-D(680mg,69%),为灰白色固体。SZ-D:1HNMR(500MHz,CDCl3)δ(ppm):1.02(3H,d),0.91(3H,s)。合成化合物SZ-E。向无水甲醇(100mL)中的化合物SZ-D(3.24g,11.24mmol)溶液添加对甲苯磺酸一水合物(193mg,1.12mmol)。于70℃将溶液加热3小时。通过添加饱和Na2CO3溶液(10mL)淬灭反应。在真空中除去大部分甲醇溶剂。然后,用乙酸乙酯(200mL)稀释残留物。用饱和Na2CO3溶液(2×100mL)清洗所得的溶液。用乙酸乙酯(50mL)提取组合的水层。用卤水(100mL)清洗组合的有机提取物,通过硫酸镁干燥并且在真空中浓缩。通过快速层析纯化残留物(洗脱剂:石油醚/EtOAc=15:1,添加0.1%NEt3)以提供产物SZ-E(1.76g,47%),为灰白色固体。此外,还回收起始化合物SZ-E(1.34g)。基于回收起始材料的收率是93%。SZ-E:1HNMR(500MHz,d6-丙酮)δ(ppm):3.080(3H,s),3.076(3H,s),2.37(1H,dd),1.98(1H,dd),0.91(3H,d),0.85(3H,s)。合成化合物SZ-F。向无水THF(25mL)中的乙基三苯基溴化膦(6.67g,17.96mmol)的悬浮液添加t-BuOK(2.01g,17.96mmol)。将溶液转变为红色颜色,然后在70℃加热2小时。然后,按一份添加化合物SZ-E(2.00g,5.99mmol)。在70℃加热溶液过夜。通过添加水(10mL)淬灭反应。用乙酸乙酯(200mL)稀释混合物,并且用卤水(2×100mL)清洗所得的溶液,通过硫酸镁干燥并且在真空中浓缩。在没有进一步纯化的情况下将粗制产物SZ-F直接用于下一步骤。合成化合物SZ-G:向THF(50mL)中的粗制产物SZ-F(2.25g,6.50mmol,理论量)添加4MHCl(2mL)。在周围温度将溶液搅拌1小时。用乙酸乙酯(300mL)稀释混合物,并且用饱和Na2CO3溶液(2×100mL)清洗所得的溶液。用EtOAc(100mL)提取组合的水层。用卤水(100mL)清洗组合的有机提取物,通过硫酸镁干燥并且在真空中浓缩。通过快速层析纯化残留物(洗脱剂:石油醚/EtOAc=20:1)以提供期望的产物SZ-G,1.78g(5.94mmol,91%收率)。SZ-G:1HNMR(500MHz,CDCl3)δ(ppm):5.13(1H,qt),1.66(3H,dt),1.02(3H,d),0.91(3H,s)。合成化合物SZ-H:向无水DMSO(30mL)中的三甲基碘化亚砜(6.53g,29.70mmol)溶液添加氢化钠(60%wt,1.19mg,29.70mmol)。在25℃搅拌混合物1小时。然后,添加无水THF(10mL)中的粗制化合物SZ-G(2.05g,被一些PPh3污染,理论量,1.78g,5.94mmol)的溶液。在25℃将混合物搅拌过夜。通过添加水(5mL)淬灭反应。用乙酸乙酯(300mL)稀释混合物,并且用水(2×100mL),接着用卤水(100mL)清洗所得的溶液,通过硫酸镁干燥并且在真空中浓缩。在没有进一步纯化的情况下将粗制产物SZ-H直接用于下一步骤。合成化合物SZ-I:向无水THF(30mL)中的粗制反应物SZ-H(理论量,1.21g,3.85mmol)溶液按份添加氢化铝锂(lithiumalumiumhydride)(731mg,19.25mmol)。在25℃将悬浮液搅拌1小时。然后,通过添加EtOAc(5mL),接着添加水(5mL)淬灭反应。过滤灰白色固体,并且用EtOAc(5×100mL)彻底清洗。用卤水(200mL)清洗组合的滤液,通过硫酸镁干燥并且在真空中浓缩。通过快速层析纯化残留物(洗脱剂:石油醚/EtOAc=15:1)以提供产物SZ-I(560mg,1.78mmol,2步总收率,30%),为灰白色固体。SZ-I:1HNMR(500MHz,CDCl3)δ(ppm):5.11(1H,qt),2.05(1H,s),1.56(3H,s),1.17(3H,s),0.91(3H,d),0.88(3H,s)。合成化合物SZ-J。向无水THF(20mL)中的反应物SZ-I(320mg,1.013mmol)溶液添加BH3.THF(1.0M,5.07mL,5.065mmol),在25℃将此溶液搅拌过夜,然后,通过添加水(4mL)淬灭反应。添加2M水性NaOH溶液(8mL),接着添加30%H2O2(8mL)。在室温将混合物搅拌1小时。用EtOAc(200mL)稀释混合物,并且用卤水(2×100mL)清洗所得的溶液,通过硫酸镁干燥并且在真空中浓缩。在没有进一步纯化的情况下将粗制产物SZ-J直接用于下一步骤。合成化合物SZ-K。向二氯甲烷(30mL)中的粗制化合物SZ-J(320mg,1.013mmol)溶液按份添加重铬酸吡啶(PDC)(1.14mg,3.039mmol)。在25℃搅拌溶液过夜。然后,将混合物过滤通过硅胶的短垫并且用二氯甲烷(3×50mL)清洗硅胶。将所有滤液组合并且在真空中浓缩。通过快速层析纯化残留物(洗脱剂:石油醚/EtOAc=6:1)以提供产物SZ-K(140mg,0.422mmol,收率42%,2steps),为灰白色固体。SZ-K:1HNMR(500MHz,CDCl3)δ(ppm):2.54(1H,t),2.12(3H,s),1.99(1H,td),1.82-1.86(1H,m),1.18(3H,s),0.92(3H,d),0.61(3H,s)。SZ-K:13CNMR(100MHz,CDCl3)δ(ppm):209.79,71.09,63.94,55.87,47.94,47.78,46.97,44.35,41.16,40.20,39.04,37.93,34.48,33.13,31.55,30.91,28.45,25.80,24.20,22.73,15.15,13.43。合成化合物SZ。向甲醇(10mL)中的化合物SZ-K(100mg,0.301mmol)溶液添加48%氢溴酸(152mg,0.903mmol),接着是溴(241mg,0.077mL,1.505mmol)。在25℃将溶液加热2小时。然后,将混合物倒入冷却水(50mL)。用乙酸乙酯(2×50mL)提取所得的固体。用卤水(50mL)清洗组合的有机提取物,通过硫酸镁干燥并且在真空中浓缩。在没有进一步纯化的情况下将粗制产物SZ直接用于下一步骤。实施例61:合成化合物SZ-1和SZ-2。向无水THF(6mL)中的粗制化合物SZ(80mg,0.195mmol)溶液添加1,2,3-三唑(40.4mg,0.585mmol),接着是碳酸钾(80.9mg,0.585mmol)。在50℃将溶液加热过夜。然后,用EtOAc(100mL)稀释溶液。用卤水(2×50mL)清洗所得的溶液,通过硫酸镁干燥并且在真空中浓缩。通过反相prep-HPLC纯化粗制产物以提供产物SZ-1(15mg,19%)和产物SZ-2(6mg,7.7%),为灰白色固体。SZ-1:1HNMR(500MHz,CDCl3)δ(ppm):7.77(1H,s),7.65(1H,s),5.28(1H,AB),5.14(1H,AB),2.66(1H,t),1.18(3H,s),0.92(3H,d),0.68(3H,s).SZ-2:1HNMR(500MHz,CDCl3)δ(ppm):7.69(2H,s),5.25(1H,AB),5.23(1H,AB),2.60(1H,t),1.18(3H,s),0.92(3H,d),0.71(3H,s)。实施例62:合成SS和SS中间体。合成化合物SS-A1和SS-A2。在-78℃在N2下向THF(25mL)和HMPA(0.5mL)中的化合物SB-F(800mg,2.79mmol)和PhSO2CF2H(540mg,2.79mmol)溶液逐滴添加LHMDS(4mL,在THF中的1M)。在–78℃搅拌2h后,用饱和水性NH4Cl溶液(10mL)淬灭反应混合物并且允许加热到室温然后用Et2O(20mL×3)提取。用卤水清洗组合的有机层,通过硫酸钠干燥,过滤并且浓缩。通过硅胶柱层析纯化残留物(石油醚/乙酸乙酯=10/1)以给出化合物SS-A1和SS-A2的混合物(650mg)。通过手性-HPLC进一步纯化混合物以提供化合物SS-A1(250mg,t=3.29min)和SS-A2(230mg,t=3.89min)。合成化合物SS-B2。在–20℃在N2下向无水甲醇(5mL)中的化合物SS-A2(230mg,0.489mmol)和无水Na2HPO4(150mg)溶液添加Na/Hg汞齐(700mg)。在–20℃至0℃搅拌1h后,弃去甲醇溶液,并且用Et2O(5x3mL)清洗固体残留物。在真空下除去组合的有机相,并且添加20ml卤水,接着用Et2O提取。用MgSO4干燥组合的醚相,过滤并且浓缩。通过硅胶层析(PE/EA=10/1)纯化粗制产物以给出化合物SS-B2(120mg,73%)。1HNMR(400MHz,CD3COCD3),δ(ppm),6.02-5.88(t,1H),5.13-5.08(m,1H),0.92(s,3H)。合成化合物SS-C2。向干THF(5mL)中的化合物SS-B2(120mg,0.355mmol)溶液添加硼烷-四氢呋喃复合物(1.20mL;THF中的1.0M溶液)。在室温搅拌1小时后,在冰浴中冷却反应混合物,然后用10%水性NaOH(1mL),接着用H2O2的30%水溶液(1.2mL)缓慢淬灭。允许混合物在室温搅拌1小时。然后,用EtOAc(3x10mL)提取。用10%水性Na2S2O3(10mL),卤水(10mL)清洗组合的有机层,通过MgSO4干燥,过滤并浓缩以提供化合物SS-C2(180mg,粗制)。在没有进一步纯化的情况下将粗制产物用于下一步骤。合成化合物SS-D2。向10mL湿二氯甲烷(二氯甲烷已经与几毫升H2O一起摇动,然后与水层分开)中的化合物SS-C2(180mg,粗制)溶液添加Dess-Martin过碘化物(380mg,0.896mmol)。在室温搅拌24h后,用二氯甲烷(3x10mL)提取反应混合物。用10%水性Na2S2O3(10mL),卤水(10mL)清洗组合的有机层,通过MgSO4干燥,过滤并且浓缩。通过硅胶上层析(石油醚/乙酸乙酯=1:5)纯化残留物以提供化合物SS-D2(70mg,对于两步为55.7%),为灰白色固体。1HNMR(400MHz,CDCl3),δ(ppm),5.90-5.61(t,1H),2.48-2.43(m,1H),2.10(s,3H),0.55(s,3H)。合成化合物SS。向MeOH(5mL)中的化合物SS-D2(50mg,0.14mmol)溶液添加2滴HBr(48%),接着是溴(100mg,0.62mmol)。在室温搅拌1h后,将反应混合物倒入冰水中,然后用乙酸乙酯(15mLx3)提取。用卤水(20mL)清洗组合的有机层,通过MgSO4干燥,过滤并浓缩以给出化合物SS(72mg,粗制)。在没有进一步纯化的情况下将粗制产物用于下一步骤。实施例63:合成化合物SS-1。向THF(10mL)中的K2CO3(126mg,0.92mmol)悬浮液添加1,2,3-1H-三唑(22.4mg,0.92mmol)和化合物SS(200mg,0.46mmol)。在室温搅拌15h后,将反应混合物倒入5mLH2O中并且用EtOAc(2×10mL)提取。用卤水清洗组合的有机层(2×10mL),通过硫酸钠干燥,过滤并且在真空中浓缩。通过反相prep-HPLC纯化残留物以提供SS-1,为灰白色固体(53.8mg,0.13mmol,27.7%)。SS-1:1HNMR(400MHz,CDCl3)δ(ppm):7.76(d,1H),7.64(d,1H),5.82(t,1H),5.25(AB,1H),5.13(AB,1H),2.65(t,1H),0.69(s,3H).LCMS:Rt=2.01min.m/z=422.3[M+H]+。实施例64:合成SN和SN中间体。合成化合物SN-B。向吡啶(30mL)中的反应物SN-A(10.0g,36.44mmol)溶液添加添加乙酸酐(5.0mL,52.89mmol)。在60℃将混合物搅拌过夜。然后,将溶液导入冰水(200mL)中。过滤白色沉淀物,并且溶解于乙酸乙酯(300mL)。用饱和CuSO4.5H2O溶液(2×200mL)清洗所得的溶液以除去残留的吡啶。用卤水(200mL)进一步清洗有机层,通过硫酸镁干燥并且在真空中浓缩。通过快速层析纯化残留物(洗脱剂:石油醚/乙酸乙酯=4:1)以提供产物SN-B(11.125g,35.16mmol,收率=96%),为灰白色固体。SN-B:1HNMR(500MHz,CDCl3)δ(ppm):5.83(1H,s),4.62(1H,dd),2.05(3H,s),0.86(3H,s)。合成化合物SN-C。在-78℃向THF(150mL)中的反应物SN-B(4.68g,14.79mmol)溶液添加LiHMDS(1.0MinTHF溶液,17.74mL,17.74mmol)。在-78℃将溶液搅拌30分钟。然后,添加HMPA(3.09mL,17.74mmol)。在-78℃将溶液再搅拌30分钟。然后,添加碘甲烷(2.76mL,44.37mmol)。在-78℃将溶液进一步搅拌2小时,并且加热到室温,并且搅拌1小时。通过添加水(10mL)淬灭反应。在真空中除去大部分THF溶剂。然后,用乙酸乙酯(300mL)稀释残留物,并且用卤水(2×200mL)清洗所得的溶液,通过硫酸镁干燥。在真空中除去溶剂提供了粗制产物SN-C(4.50g,13.62mmol,收率=92%),为稠油。在没有进一步纯化的情况下将粗制产物用于下一步骤。SN-C:1HNMR(500MHz,CDCl3)δ(ppm):5.75(1H,s),4.62(1H,t),2.05(3H,s),1.10(3H,d),0.86(3H,s)。合成化合物SN-D1&SN-D2。向甲醇(100mL)和水(20mL)中的粗制反应物SN-C(11.62g,35.16mmol,理论量)溶液添加氢氧化钠(2.81g,70.32mmol)。在60℃将溶液加热1小时。然后,在真空中除去大部分甲醇溶剂。通过2MHCl将残留的溶液酸化为pH5-6。用乙酸乙酯(3×100mL)提取水层。用卤水(200mL)清洗组合的有机提取物,通过硫酸镁干燥并且在真空中浓缩。通过快速层析纯化残留物(洗脱剂:石油醚/乙酸乙酯=5:1)以提供纯产物SN-D1(2.354g,8.162mmol,收率=23%)和纯产物SN-D2(5.306g,18.40mmol,收率=50%),为灰白色固体。SN-D1:1HNMR(500MHz,CDCl3)δ(ppm):5.81(1H,s),3.67(1H,t),1.11(3H,d),0.81(3H,s).SN-D2:1HNMR(500MHz,CDCl3)δ(ppm):5.74(1H,s),3.67(1H,t),1.11(3H,d),0.81(3H,s)。合成化合物SN-E。在-78℃向液体氨(200mL)添加锂(1.80g,260mmol)。然后,将液体转变为深蓝色。然后,将t-BuOH(1.0mL,10.40mmol)和THF(100mL)中的反应物SN-D1(3.0g,10.40mmol)溶液添加到Li-氨溶液。在-78℃将混合物搅拌4小时。然后,添加NH4Cl固体(20g)以淬灭反应。将混合物从深蓝色转变为白色。让混合物加热到室温,并且在通风柜中蒸发氨过夜。对残留物添加水(300mL)。通过浓HCl将混合物酸化为pH6-7。然后,添加乙酸乙酯(300mL)。用乙酸乙酯(2×100mL)进一步提取分离的水层。用卤水(300mL)清洗组合的有机提取物,通过硫酸镁干燥并且在真空中浓缩。在没有进一步纯化的情况下将粗制产物SN-E直接用于下一步骤。合成化合物SN-F。向二氯甲烷(60mL)中的粗制反应物SN-E(1.749g,6.022mmol)溶液添加重铬酸吡啶(PDC)(3.398g,9.033mmol)。在室温将混合物搅拌过夜。将溶液过滤通过硅藻土的短垫。用CH2Cl2(3×50mL)清洗硅藻土。在真空中浓缩组合的CH2Cl2溶液。通过快速层析纯化残留物(洗脱剂:石油醚/乙酸乙酯=5:1)以提供产物SN-F(1.298g,4.50mmol,收率=75%),为灰白色固体。SN-F:1HNMR(400MHz,CDCl3)δ(ppm):1.02(3H,d),0.91(3H,s)。合成化合物SN-G。向无水甲醇(50mL)中的反应物SN-F(1.948g,6.754mmol)溶液添加对甲苯磺酸一水合物(128mg,0.6754mmol)。在70℃将溶液加热3小时。通过添加饱和Na2CO3溶液(10mL)淬灭反应。在真空中除去大部分甲醇溶剂。然后,用乙酸乙酯(200mL)稀释残留物。用饱和的Na2CO3溶液(2×100mL)清洗所得的溶液。用乙酸乙酯(50mL)提取组合的水层。用卤水(100mL)清洗组合的有机提取物,通过硫酸镁干燥并且在真空中浓缩。通过快速层析纯化残留物(洗脱剂:石油醚/乙酸乙酯=10:1,添加0.1%NEt3)以提供产物SN-G(652mg,1.949mmol,收率=29%),为灰白色固体。此外,还回收起始材料(1.338g)。因此,基于回收的起始材料的收率是92%。SN-G:1HNMR(500MHz,d6-丙酮)δ(ppm):3.079(3H,s),3.075(3H,s),2.38(1H,dd),1.98(1H,dd),0.91(3H,d,J=7.2Hz),0.85(3H,s)。合成化合物SN-H。向无水THF(20mL)中的乙基三苯基溴化膦(8.795g,23.69mmol)的溶液添加t-BuOK(2.658g,23.69mmol)。然后,溶液变为微红颜色,并且在70℃加热2小时。然后,按一份添加反应物SN-G(1.642g,4.909mmol)。在70℃加热溶液过夜。通过添加水(10mL)淬灭反应。用乙酸乙酯(200mL)稀释混合物,并且用卤水(2×100mL)清洗所得的溶液,通过硫酸镁干燥并且在真空中浓缩。在没有进一步纯化的情况下将粗制产物SN-H直接用于下一步骤。合成化合物SN-I。向THF(30mL)中的粗制产物SN-H(1.702g,4.909mmol,理论量)添加2MHCl(3mL)。在周围温度将溶液搅拌1小时。用乙酸乙酯(300mL)稀释混合物,并且用饱和Na2CO3溶液(2×100mL)清洗所得的溶液。用乙酸乙酯(100mL)提取组合的水层。用卤水(100mL)清洗组合的有机提取物,通过硫酸镁干燥并且在真空中浓缩。通过快速层析纯化残留物(洗脱剂:石油醚/乙酸乙酯=100:3)以提供粗制产物SN-I(1.746g),为灰白色固体,其被一些未分离的PPh3污染。通过合并1HNMR谱判断,期望产物与PPh3的比率是3:1,因此期望的产物SN-I量是1.354g(4.506mmol),收率是92%。SN-I:1HNMR(500MHz,CDCl3)δ(ppm):5.13(1H,qt),1.66(3H,dt),1.02(3H,d),0.91(3H,s)。合成化合物SN-J。向无水DMSO(30mL)中的三甲基碘化亚砜(5.213g,23.69mmol)溶液添加氢化钠(60%wt,948mg,23.69mmol)。在25℃将混合物搅拌1小时。然后,添加无水THF(10mL)中的粗制反应物(1.746g,被一些残留PPh3污染,理论量,1.354g,4.506mmol)溶液。在25℃将混合物搅拌过夜。通过添加水(5mL)淬灭反应。用乙酸乙酯(300mL)稀释混合物,并且用水(2×100mL),接着用卤水(100mL)清洗所得的溶液,通过硫酸镁干燥并且在真空中浓缩。在没有进一步纯化的情况下将粗制产物SN-J直接用于下一步骤。合成化合物SN-K。向无水THF(30mL)中的粗制反应物SN-J(理论量,1.417g,4.506mmol)溶液按份添加氢化铝锂(342mg,9.012mmol)。在25℃将悬浮液搅拌1小时。然后,通过添加乙酸乙酯(5mL),接着添加水(5mL)淬灭反应。将灰白色固体过滤,并且用乙酸乙酯(5×100mL)彻底清洗。用卤水(200mL)清洗组合的滤液,通过硫酸镁干燥并且在真空中浓缩。通过快速层析纯化残留物(洗脱剂:石油醚/乙酸乙酯=20:1)以提供产物SN-K(458mg,1.447mmol,2步总收率=32%),为灰白色固体。合成化合物SN-L。向无水THF(15mL)中的反应物SN-K(458mg,1.447mmol)溶液添加BH3.THF(1.0M,7.23mL,7.23mmol),在25℃搅拌溶液过夜。然后,通过添加水(5mL)淬灭反应。添加2MNaOH溶液(10mL),接着添加30%H2O2(10mL)。在室温将混合物搅拌1小时。用乙酸乙酯(200mL)稀释混合物,并且用卤水(2×100mL)清洗所得的溶液,通过硫酸镁干燥并且在真空中浓缩。在没有进一步纯化的情况下将粗制产物直接用于下一步骤。合成化合物SN-M。向二氯甲烷(40mL)中的粗制反应物SN-L(484mg,1.447mmol,理论量)溶液按份添加重铬酸吡啶(PDC)(1633mg,4.341mmol)。在25℃搅拌溶液过夜。然后,将混合物过滤通过硅胶的短垫并且用二氯甲烷(3×50mL)清洗硅胶。将所有滤液组合并且在真空中浓缩。通过快速层析纯化残留物(洗脱剂:石油醚/乙酸乙酯=8:1)以提供产物SN-M(305mg,0.917mmol,收率=63%(2步)),为灰白色固体。SN-L:1HNMR(500MHz,CDCl3)δ(ppm):2.54(1H,t0,2.12-2.19(1H,m),2.12(3H,s),1.99(1H,td),1.80-1.86(1H,m),1.17(3H,s),0.92(3H,d),0.61(3H,s)。SN-M:13CNMR(100MHz,CDCl3)δ(ppm):209.75,71.09,63.96,55.89,47.96,47.80,47.00,44.35,41.19,40.22,39.05,37.95,34.49,33.14,31.54,30.92,28.46,25.82,24.22,22.76,15.14,13.45。合成化合物SN。向甲醇(10mL)中的反应物SN-M(100mg,0.301mmol)溶液添加48%氢溴酸(152mg,0.903mmol),接着是溴(241mg,0.077mL,1.505mmol)。在25℃将溶液加热1.5小时。然后,将混合物倒入冷却水(50mL)。用乙酸乙酯(2×50mL)提取所得的固体。用卤水(50mL)清洗组合的有机提取物,通过硫酸镁干燥并且在真空中浓缩。在没有进一步纯化的情况下将粗制产物SN直接用于下一步骤。实施例65:合成化合物SN-1和SN-2。向无水THF(6mL)中的粗制反应物SN(124mg,0.301mmol)溶液添加1,2,3-三唑(31mg,0.45mmol),接着是碳酸钾(62mg,0.45mmol)。在50℃加热溶液过夜。然后,用乙酸乙酯(100mL)稀释溶液。用卤水(2×50mL)清洗所得的溶液,通过硫酸镁干燥并且在真空中浓缩。通过反相prep-HPLC纯化粗制产物以提供产物SN-1(21mg,0.0526mmol,收率=17%)和产物SN-2(16mg,0.0400mmol,收率=13%),为灰白色固体。SN-1:HNMR(400MHz,CDCl3)δ(ppm):7.76(1H,s),7.65(1H,s),5.20(1H,AB),5.14(1H,AB),2.66(1H,t),2.21(1H,dd),1.18(3H,s),0.92(3H,d),0.68(3H,s).SN-2:1HNMR(500MHz,CDCl3)δ(ppm):7.69(2H,s),5.27(1H,AB),5.23(1H,AB),2.60(1H,t),2.20(1H,dd),1.17(3H,s),0.92(3H,d),0.71(3H,s)。实施例66:合成SU和SU中间体。合成化合物SU-B。在-78℃向NH3(液体,2.0L)添加锂(7.0g,1mol)。在将液体转变为深蓝色后,逐滴添加t-BuOH(7.4g,100mmol)和THF(20mL)中的化合物SU-A(27.0g,100mmol)溶液。在-78℃将混合物搅拌4小时。然后,添加NH4Cl固体(50g)以淬灭反应。将混合物从深蓝色转变为白色。让混合物加热到室温,并且将氨蒸发过夜。在0.5N含水HCl(50mL)中溶解残留物,并且用二氯甲烷(200mL×3)提取。用饱和NaHCO3(200mL)和卤水(200mL)清洗组合的有机层,通过硫酸镁干燥并且在真空中浓缩。通过快速层析(PE/EtOAc=4:1)纯化粗制产物以得到产物SU-B(18.98g,68.7%),为灰白色固体。SY-B:1HNMR(500MHz,CDCl3)δ(ppm):3.66(1H,t),2.29-2.27(2H,m),2.12-2.07(2H,m),1.83-1.81(2H,m),1.50(1H,s),0.77(3H,s)。合成化合物SU-C。在0℃在50mLTHF中溶解19.0g化合物SU-B(68.84mmol)样品。然后,在30min中逐滴添加THF(3M)中的70mLMeMgBr。在0℃将反应保持8h。用冰冷的水淬灭反应混合物,并且用EtOAc(200mL×3)提取。用卤水清洗组合的有机层,通过硫酸钠干燥,过滤并且浓缩。通过快速柱层析纯化白色残留物(PE/EtOAc=5:1)以给出产物SU-C(19.0g,94%),为灰白色固体。SY-C:1HNMR(500MHz,CDCl3)δ(ppm):5.78(1H,br),5.36(1H,t),3.67(1H,t),1.73(3H,s),0.77(3H,s)。合成化合物SU-D。向二氯甲烷(100mL)中的化合物SU-C(19.0g,65.07mmol)溶液添加重铬酸吡啶(PDC)(48.9g,130.14mmol)。在室温将混合物搅拌过夜。将溶液过滤通过硅藻土的短垫。用CH2Cl2(3×100mL)清洗硅藻土。在真空中浓缩组合的CH2Cl2溶液。通过快速层析纯化残留物(洗脱剂:PE/EtOAc=5:1)以提供产物SU-D(10.0g,53%),为灰白色固体。SY-D:1HNMR(500MHz,CDCl3)δ(ppm):2.44(1H,dd),2.07(1H,m),1.21(3H,s),0.87(3H,s)。合成化合物SU-E:向无水甲苯中的化合物SU-D(5.0g,17.2mmol)溶液添加硅胶(80g)上的对甲苯磺酸,在45℃下将混合物搅拌1小时。通过用PE/EtOAc(10/1)洗脱从硅胶除去不溶性副产物。在没有进一步纯化的情况下将粗制产物SU-E(3.20g,11.75mmol)用于下一步骤。合成化合物SU-F:向10mL无水二氯甲烷中的化合物SU-E(3.20g,11.75mmol)的溶液添加mCPBA(4.04g,23.50mmol),并且在室温将反应混合物搅拌过夜。然后,用CH2Cl2提取反应混合物,用NaHCO3(100mL)和卤水将组合的有机层清洗两次,通过Na2SO4干燥,并且浓缩。在没有进一步纯化的情况下将粗制产物SU-F用于下一步骤。合成化合物SU-G。向甲醇中的化合物SU-F(11.75mmol)溶液添加H2SO4(0.5mL),并且在室温将反应混合物搅拌2h。然后用CH2Cl2(200mL×3)提取反应溶液,用NaHCO3(100mL)和卤水清洗组合的有机层,通过Na2SO4干燥,并且浓缩。通过层析纯化残留物(PE/EtOAc=10:1)以提供化合物SU-G(3.30g,10.30mmol,对于两步为收率=87%),为灰白色固体。合成化合物SU-H。向无水THF(20mL)中的乙基三苯基溴化膦(11.52g,31.0mmol)溶液添加t-BuOK(3.48g,31.0mmol)。将溶液转变为微红,并且在70℃加热3小时。然后,按一份添加化合物SU-G(3.30g,10.30mmol)。在70℃将反应溶液加热过夜,然后通过添加水(10mL)淬灭。用EtOAc(200mL)稀释混合物,并且用卤水(2×100mL)清洗所得的溶液,通过硫酸镁干燥并且在真空中浓缩。在没有进一步纯化的情况下将粗制产物SU-H(1.90g)直接用于下一步骤。合成化合物SU-I。向干THF(20mL)中的化合物SU-H(1.90g,5.72mmol)溶液添加BH3-THF(18mLof1.0M溶液inTHF)。在室温搅拌1h后,在冰浴中冷却反应混合物,然后用10%水性NaOH(12mL),接着用30%H2O2(20mL)缓慢淬灭。在室温让混合物搅拌1h,然后用EA(100mL×3)提取。用10%水性Na2S2O3(50mL),卤水清洗组合的有机层,通过Na2SO4干燥,过滤并浓缩以提供粗制化合物SU-I(1.86g,5.31mmol)。在没有进一步纯化的情况下将粗制产物用于下一步骤。合成化合物SU-J。向二氯甲烷(50mL)中的粗制化合物SU-I(1.86g,5.31mmol)溶液按份添加重铬酸吡啶(PDC)(3.98g,10.62mmol)。在25℃搅拌溶液过夜。然后,将混合物过滤通过硅胶的短垫并且用二氯甲烷(3×50mL)清洗硅胶。将所有滤液组合并且在真空中浓缩。通过快速层析纯化残留物(洗脱剂:PE/EtOAc=10:1)以提供产物SU-J(1.20g,3.45mmol,65%),为灰白色固体。SU-J:1HNMR(500MHz,CDCl3)δ(ppm):3.33(3H,s),3.04(1H,s),2.53(1H,t),2.12(3H,s),1.26(3H,s),0.62(3H,s)合成化合物SU。向甲醇(10mL)中的反应物SU-J(100mg,0.287mmol)溶液添加48%HBr(152mg,0.903mmol),接着是溴(0.08mL,1.505mmol)。在25℃将溶液加热1.5小时。然后,将混合物倒入冷却水(50mL)。用乙酸乙酯(2×50mL)提取所得的固体。用卤水(50mL)清洗组合的有机提取物,通过硫酸镁干燥并且在真空中浓缩。在没有进一步纯化的情况下将粗制产物SU直接用于下一步骤。实施例67:合成SU-1和SU-2。向无水THF(6mL)中的粗制化合物SU(100mg,0.243mmol)溶液添加1,2,3-三唑(34mg,0.50mmol),接着是碳酸钾(70mg,0.50mmol)。在50℃将溶液加热过夜。然后,用EtOAc(100mL)稀释溶液。用卤水(2×50mL)清洗所得的溶液,通过硫酸镁干燥并且在真空中浓缩。通过反相prep-HPLC纯化粗制产物以提供产物SU-1(35mg,0.084mmol,收率=34%)和产物SU-2(20mg,0.048mmol,20%),为灰白色固体。SU-1:1HNMR(500MHz,CDCl3)δ(ppm):7.76(1H,s),7.65(1H,s),5.27(1H,AB),5.14(1H,AB),3.34(3H,s),3.04(1H,s),2.65(1H,t),1.24(3H,s),0.68(3H,s).SU-2:1HNMR(500MHz,CDCl3)δ(ppm):7.68(2H,s),5.26(1H,AB),5.22(1H,AB),3.33(3H,s),3.04(1H,s),2.59(1H,t),1.24(3H,s),0.72(3H,s)。实施例68:合成SY和SY中间体。合成化合物SY-B。在-78℃向NH3(液体,2.0L)添加锂(7.0g,1mol)。在液体转变为深蓝色后,逐滴添加t-BuOH(7.4g,100mmol)和THF(20mL)中的反应物SY-A(27.0g,100mmol)溶液。在-78℃将混合物搅拌4小时,然后添加NH4Cl固体(50g)以淬灭反应。然后,混合物从深蓝色转变为白色。让混合物加热到室温,并且在通风柜中蒸发氨过夜。在0.5NHCl(50mL)中溶解残留物,并且用二氯甲烷(200mL×3)提取。用饱和NaHCO3(200mL)和卤水(200mL)清洗组合的有机层,通过硫酸镁干燥并且在真空中浓缩。通过快速层析(PE/EA=4:1)纯化粗制产物以得到产物SY-B(18.98g,68.76mmol,收率=68.7%),为灰白色固体。SY-B:1HNMR(500MHz,CDCl3)δ(ppm):3.66(1H,t),2.29-2.27(2H,m),2.12-2.07(2H,m),1.83-1.81(2H,m),1.50(1H,s),0.77(3H,s)。合成化合物SY-C。在0℃在50mLTHF中溶解19.0g化合物SY-B(68.84mmol)。然后,在30分钟里逐滴添加THF(3M)中的70mLMeMgBr,然后在0℃将反应保持8h。用冰冷的水淬灭反应混合物,并且用EA(200mL×3)提取。用卤水清洗组合的有机层,通过硫酸钠干燥,过滤并且浓缩。通过快速柱层析纯化白色残留物(PE/EA=5:1)以得到产物SY-C(19.0g,65.07mmol收率=94%),为灰白色固体。SY-C:1HNMR(500MHz,CDCl3)δ(ppm):5.78(1H,br),5.36(1H,t),3.67(1H,t),1.73(3H,s),0.77(3H,s)。合成化合物SY-D。在室温向二氯甲烷(100mL)中的反应物SY-C(19.0g,65.07mmol)溶液添加重铬酸吡啶(PDC)(48.9g,130.14mmol),并且在室温将混合物搅拌过夜。将溶液过滤通过硅藻土的短垫。用CH2Cl2(3×100mL)清洗硅藻土。在真空中浓缩组合的CH2Cl2溶液。通过快速层析纯化残留物(洗脱剂:PE/EA=5:1)以提供产物SY-D(10.0g,34.48mmol,收率=53%),为灰白色固体。SY-D:1HNMR(500MHz,CDCl3)δ(ppm):2.44(1H,dd),2.07(1H,m),1.21(3H,s),0.87(3H,s)。合成化合物SY-E。向无水甲苯(100mL)中的反应物SY-D(5.0g,17.2mmol)溶液快速添加硅胶(80g)上的对甲苯磺酸,在45℃将混合物搅拌1小时。通过用(PE/EA=30:1)洗脱从硅胶中除去产物。在没有进一步纯化的情况下将粗制产物SY-E(3.20g,11.75mmol)用于下一步骤。合成化合物SY-F。向10mL无水二氯甲烷中的SY-E(3.20g,11.75mmol)溶液添加mCPBA(4.04g,23.50mmol),并且在室温将反应混合物搅拌过夜。然后,用CH2Cl2(2x100mL)提取溶液,并且用NaHCO3(100mL)和卤水将组合的有机层清洗两次,通过Na2SO4干燥,并且浓缩。在没有进一步纯化的情况下将粗制产物SY-E用于下一步骤。合成化合物SY-G。向乙醇(50mL)中的SY-F(900mg,3.12mmol)溶液添加浓H2SO4(0.5mL),并且在室温将反应混合物搅拌2h。只要TLC指示完全转化,用CH2Cl2(200mL×3)提取溶液,用NaHCO3(100mL)和卤水清洗组合的有机层,通过Na2SO4干燥,并且浓缩。通过层析纯化残留物(PE/EA=10:1)以提供化合物SY-G(600mg,1.80mmol,对于两步为收率=57.6%),为灰白色固体。合成化合物SY-H。向无水THF(10mL)中的乙基三苯基溴化膦(1.99g,5.96mmol)溶液添加t-BuOK(500mg,4.48mmol)。将溶液转变为微红,并且在70℃加热3小时。然后,按一份添加反应物SY-G600mg,1.79mmol)。在70℃将溶液加热过夜。通过添加水(5mL)淬灭反应。用乙酸乙酯(100mL)稀释混合物,并且用卤水(2×50mL)清洗所得的溶液,通过硫酸镁干燥并且在真空中浓缩。通过快速层析纯化残留物(洗脱剂:石油醚:乙酸乙酯=20:1)以提供产物SY-H(1.55g,4.48mmol,75.2%),为灰白色固体。合成化合物SY-I。向干THF(20mL)中的化合物SY-H(1.20g,3.47mmol)溶液添加BH3-THF(THF中的18mL1.0M溶液)。在室温搅拌1h后,在冰浴中冷却反应混合物,然后用10%水性NaOH(10mL),接着用30%H2O2(15mL)缓慢淬灭。在室温让混合物搅拌1h,然后用EA(100mL×3)提取。用10%水性Na2S2O3(50mL),卤水清洗组合的有机层,通过Na2SO4干燥,过滤并浓缩以提供粗制化合物SY-I(1.12g)。在没有进一步纯化的情况下将粗制产物用于下一步骤。合成化合物SY-J。在室温向二氯甲烷(50mL)中的粗制反应物SY-I(1.12g,3.08mmol)溶液按份添加重铬酸吡啶(PDC)(3.32g,6.16mmol)。在25℃将溶液搅拌过夜,然后将混合物过滤通过硅胶的短垫并且用二氯甲烷(3×50mL)清洗硅胶。将所有滤液组合并且在真空中浓缩。通过快速层析纯化残留物(洗脱剂:PE/EA=10:1)以提供产物SY-J(0.95g,2.62mmol,收率=85.1%),为灰白色固体。SY-J:1HNMR(500MHz,CDCl3)δ(ppm):3.59(1H,m),3.36(1H,m),3.12(1H,s),2.53(1H,t),2.14(3H,s),1.23(3H,s),1.17(3H,t),0.62(3H,s)。合成化合物SY。向甲醇(5mL)中的反应物SY-J(80mg,0.221mmol)溶液添加48%氢溴酸(148mg,0.884mmol),接着是溴(241mg,0.077mL,1.505mmol)。在25℃将溶液加热1.5小时,然后,将混合物倒入冷却水(50mL)。用乙酸乙酯(2×50mL)提取所得的固体。用卤水(20mL)清洗组合的有机提取物,通过硫酸镁干燥并且在真空中浓缩。在没有进一步纯化的情况下将粗制产物SY直接用于下一步骤。实施例69:合成化合物SY-1和SY-2。向干THF(5mL)中的化合物SY(110mg,粗制)溶液添加碳酸钾(100mg)和0.3mL1H-1,2,3-三唑。在50℃将反应混合物搅拌2天,然后用EtOAc(3x10mL)提取。用卤水(10mL)清洗组合的EtOAc提取物,通过Na2SO4干燥,过滤,并且浓缩。通过制备型HPLC纯化残留物以提供标题化合物SY-1(23mg,收率=21%)和SY-2(9mg,收率=8%),为灰白色固体。SY-1:1HNMR(500MHz,CDCl3)δ(ppm):7.77(1H,s),7.66(1H,s),5.27(1H,AB),5.13(1H,AB),3.61-3.57(1H,m),3.38-3.34(1H,m),3.12(1H,s),2.64(1H),1.23(3H,s),1.16(3H,t),0.68(3H,s)。SY-2:1HNMR(500MHz,CDCl3)δ(ppm):7.68(2H,s),5.26(1H,AB),5.21(1H,AB),3.61-3.57(1H,m),3.38-3.34(1H,m),3.12(1H,s),2.59(1H,t),2.08-1.97(1H,m),1.23(3H,s),1.16(3H,t),0.72(3H,s)。实施例70:合成化合物SA-10,SA-11,SA-12,SA-13,和SA-14。步骤1。在25℃向丙酮(50mL)中的SA(4.3g,10.8mmol)溶液添加K2CO3(2.98g,21.6mmol)和2H-1,2,3-三唑-4-羧酸乙酯(2.28g,16.2mmol)。在25℃将混合物搅拌12小时。TLC显示起始材料是消失的。通过水(30mL)淬灭反应,并且用EA(30mL*2)提取。用饱和卤水(50mL)清洗组合的有机相,通过无水Na2SO4干燥,过滤并且在真空中浓缩。通过硅胶层析(100-200目硅胶,石油醚/乙酸乙酯=3/1至EA)纯化残留物以提供SA-10(2.6g,纯度:95%,收率:50%,用于递送的54mg),为白色固体,和SA-11(1.5g,纯度:95%,收率:28.7%,用于递送的21mg),为浅黄色固体。SA-10:1HNMRCDCl3Bruker_P_400MHzδ8.11(s,1H),5.35-5.22(m,2H),4.43(q,J=7.3Hz,2H),2.64-2.55(m,1H),2.24-2.03(m,2H),1.91-1.60(m,7H),1.50-1.23(m,18H),1.18-1.04(m,3H),0.71(s,3H)。LCMSRt=在2min层析中的1.081min,30-90AB,纯度98.95%,MSESI计算。对于C26H39N3O4[M-H2O+H]+440,发现440。SA-11:1HNMRCDCl3Bruker_P_400MHzδ8.17(s,1H),5.33-5.12(m,2H),4.43(q,J=7.1Hz,2H),2.71-2.61(m,1H),2.28-2.15(m,1H),2.06(d,J=12.0Hz,1H),1.90-1.70(m,6H),1.69-1.60(m,1H),1.55-1.38(m,10H),1.37-1.21(m,8H),1.18-1.02(m,3H),0.67(s,3H).LCMSRt=在2min层析中的1.000min,30-90AB,纯度96.6%,MSESI计算。对于C26H39N3O4[M-H2O+H]+440,发现440。步骤2。在25℃向EtOH(8mL)中的SA-10(300mg,655umol)溶液添加LiOH.H2O(137mg,3.27mmol)。在25℃将混合物搅拌16小时。LCMS显示了反应是完全的。将反应倒入水(20mL)中,并且用HCl(2M)酸化到pH3-4,用EA(50mL*2)提取。在真空中浓缩组合的有机层。通过prep-HPLC(0.05%HCl-ACN)纯化残留物以提供SA-12(90mg,纯度:100%,收率:32%,用于递送的28mg)和副产物(13mg,纯度:100%,收率:4.62%),为白色固体。SA-12:1HNMRDMSOBruker_P_400MHzδ8.24(s,1H),5.76-5.39(m,2H),4.27(s,1H),2.78(s,1H),2.05(d,J=11.5Hz,2H),1.76-1.59(m,8H),1.49-1.23(m,10H),1.16-1.04(m,8H),0.61(s,3H).LCMSRt=在2分钟层析中的0.913min,30-90AB,纯度96.6%,MSESI计算。对于C24H35N3O4[M+Na]+452,发现452。步骤3。在25℃向DCM(15mL)中的SA-12(300mg,698umol)溶液添加HATU(395mg,1.04mmol),DIEA(224mg,1.74mmol)。在25℃将混合物搅拌15min。将氨水合物(ammoniahydrate)(0.3mL,水中的26%)添加到溶液。在25℃将混合物搅拌30min。LCMS显示了反应是完全的。浓缩混合物。通过prep-HPLC(0.05%HCl-ACN)纯化残留物以提供SA-13(120mg,纯度:100%,收率:40.1%),为白色固体。SA-13:1HNMRDMSOBruker_A_400MHzδ8.13(s,1H),7.81(s,1H),7.54(s,1H),5.63-5.33(m,2H),4.24(s,1H),2.79-2.71(m,1H),2.08-1.97(m,2H),1.78-0.94(m,25H),0.60(s,3H)。LCMSRt=在2min层析中的0.878min,30-90AB,纯度100%,MSESI计算。对于C24H36N4O3[M-H2O+H]+411,发现411。步骤4。在25℃向DCM(4mL)中的SA-13(82mg,191umol)溶液添加TFAA(120mg,573umol),吡啶(60.3mg,764umol)。在25℃将混合物搅拌16小时。LCMS显示反应是完全的。浓缩混合物,并且通过prep-HPLC(0.1%TFA-ACN)纯化残留物以提供SA-14(35mg,纯度:98.8%,收率:44%),为白色固体。SA-14:1HNMRCDCl3Bruker_P_400MHzδ8.04-8.00(m,1H),5.35-5.24(m,2H),2.67-2.60(m,1H),2.26-2.04(m,3H),1.96-1.86(m,3H),1.83-1.72(m,6H),1.69-1.62(m,4H),1.58-1.06(m,11H),0.71(s,3H).LCMSRt=在2min层析中的1.313min,30-90AB,纯度98.8%,MSESI计算。对于C24H34N4O2[M-H2O+H]393,发现393。实施例71:合成化合物SA-17,SA-18,和SA-20。步骤6。在25℃向EtOH(5mL)中的SA-11(200mg,437umol)溶液添加LiOH.H2O(91.4mg,2.18mmol)。在25℃将混合物搅拌16小时。LCMS显示反应是完全的。将反应倒入水(20mL)中,并且用HCl(2M)酸化为pH3-4,用EA(50mL*2)提取。在真空中浓缩组合的有机层。通过prep-HPLC(0.05%HCl-ACN)纯化残留物以提供SA-17(86mg,纯度:100%,收率:45.9%,用于递送的26mg)和副产物(12mg,纯度:100%,收率:6.41%),为白色固体。SA-17:1HNMRDMSOBruker_N_400MHzδ8.58(s,1H),5.62-5.33(m,2H),4.28(s,1H),2.81(t,J=8.7Hz,1H),2.14-2.00(m,2H),1.81-1.61(m,7H),1.54-1.20(m,10H),1.19-0.97(m,8H),0.61(s,3H).LCMSRt=在2min层析中的0.867min,30-90AB,纯度100%,MSESI计算。对于C24H35N3O4[M-H2O+H]412,发现412。步骤7。在25℃向DCM(10mL)中的SA-17(200mg,465umol)溶液添加HATU(264mg,697umol),DIEA(149mg,1.16mmol)。在25℃将混合物搅拌15min。将甲胺水合物(0.2mL,在水中的26%)添加到溶液。在25℃将混合物搅拌30min。LCMS显示反应是完全的。浓缩混合物。通过prep-HPLC(0.05%HCl-ACN)纯化残留物以提供SA-18(55mg,纯度:99.5%,收率:27.4%,用于递送的10mg),为白色固体。SA-18:1HNMRDMSOBruker_A_400MHzδ8.40(s,1H),7.86(s,1H),7.47(s,1H),5.59-5.31(m,2H),4.24(s,1H),2.83-2.74(m,1H),2.12-2.00(m,2H),1.78-0.96(m,23H),0.59(s,3H).LCMSRt=在2min层析中的0.877min,30-90AB,纯度100%,MSESI计算。对于C24H36N4O3[M-H2O+H]+411,发现411。步骤8。在25℃向DCM(4mL)中的SA-18(45mg,105umol)溶液添加TFAA(66.1mg,315umol),吡啶(33.1mg,420umol)。在25℃将混合物搅拌16小时。LCMS显示了反应是完全的。浓缩混合物,并且通过prep-HPLC纯化残留物以提供SA-20(6.5mg,纯度:98.89%,收率:14.8%),为白色固体。SA-20:1HNMRCDCl3Bruker_P_400MHzδ8.13(s,1H),5.37-5.13(m,2H),2.72-2.65(m,1H),2.28-2.03(m,3H),1.97-1.85(m,3H),1.84-1.73(m,5H),1.70-1.64(m,4H),1.60-1.23(m,9H),1.20-1.07(m,3H),0.67(s,3H).LCMSRt=在2min层析中的1.263min,30-90AB,纯度98.89%,MSESI计算。对于C24H34N4O2[M-H2O+H]+393,发现393。测定方法可以使用多种体外和体内测定来评价本发明提供的化合物;下面描述其实例。TBPS结合的类固醇抑制已经描述了在5μMGABA存在下的使用大鼠脑皮质膜的[35S]-二环硫代磷酸叔丁酯([35S]-t-Butylbicyclophosphorothionate)(TBPS)结合测定(Gee等,J.Pharmacol.Exp.Ther.1987,241,346-353;Hawkinson等,Mol.Pharmacol.1994,46,977-985;Lewin,A.H等,Mol.Pharmacol.1989,35,189-194)。简而言之,在将二氧化碳麻醉的Sprague-Dawley大鼠(200-250g)去头之后快速地取出皮质。使用玻璃/特氟龙匀浆器,将皮质在10体积的冰冷的0.32M蔗糖中匀浆化,并在4℃以1500xg离心10分钟。将得到的上清液在4℃以10,000xg离心20分钟,获得P2片状沉淀物(pellets)。将P2片状沉淀物再悬浮于200mMNaCl/50mM磷酸Na-KpH7.4缓冲液中,并在4℃以10,000xg离心10分钟。重复该洗涤过程两次,并将片状沉淀物再悬浮于10体积的缓冲液中。在5μMGABA的存在下,使用3nM[35S]-TBPS和溶解在二甲亚砜(DMSO)中的测试药物的5μL等分试样(最终0.5%),将膜悬浮液的等分试样(100μL)培养。用缓冲液使培养液达到1.0mL的最终体积。在2μM未标记的TBPS的存在下测定非特异性结合,且范围从15%到25%。在室温培养90分钟之后,使用细胞收集器(Brandel),过滤经过玻璃纤维过滤器(SchleicherandSchuellNo.32)终止测定,并用冰冷的缓冲液冲洗三次。通过液体闪烁光谱法测定过滤器结合的放射性。使用Prism(GraphPad),进行各平均浓度的各药物的全部数据的非线性曲线拟合。如果通过F检验,平方和显著较低,则将数据拟合为部分抑制模型,而不是完全抑制模型。类似地,如果通过F试验,平方和显著较低,则将数据拟合为双组分(twocomponent)抑制模型,而不是单组分抑制模型。使用全部数据所使用的相同模型,为单独的实验确定产生特异性结合的50%抑制(IC50)和最大抑制程度(Imax)的测试化合物的浓度,然后计算单独的实验的平均值+SEM.。印防己毒素(Picrotoxin)充当这些研究的阳性对照,这是因为已经证明它可以很好地抑制TBPS结合。筛选或可筛选多种化合物,测定它们作为[35S]-TBPS体外结合的调节剂的潜力。这些测定根据或可根据以上讨论的操作进行。重组α1β2γ2和α4β3δGABAA受体的膜片钳电生理学使用细胞电生理学,测量在异源细胞系中我们的GABAA受体调节剂的药理学性能。在次最高激动剂量(GABAEC20=2μM)下检验每个化合物影响GABA介导的电流的能力。用GABA受体的α1β2γ2亚单位稳定地转染LTK细胞,并通过Lipofecatamine方法,用α4β3δ亚单位短暂地转染CHO细胞。使细胞传代,融合度为大约50-80%,而后接种到35mm无菌培养皿上,无菌培养皿中含有2ml完全培养基,不含抗生素或抑霉物质。使细胞的融合簇进行电结合(Pritchett等,Science,1988,242,1306-1308)。因为远端细胞的反应不是充分的电压钳,并且因为结合程度的不确定性(Verdoorn等,Neuron1990,4,919-928),所以,将细胞在能够记录单细胞(看不到与其它细胞的连接)的密度下培养。利用HEKAEPC-10放大器,使用PatchMaster软件,或使用高通量QPatch平台(Sophion),测定总细胞电流。所有实验的槽液中含有(mM):NaCl:137mM,KCl:4mM,CaCl2:1.8mM,MgCl2:1mM,HEPES:10mM,D-葡萄糖:10mM,pH(NaOH)7.4。在某些情况下,还加入0.005%cremophor。胞内(吸液管)溶液含有:KCl:130mM,MgCl2:1mM,Mg-ATP:5mM,HEPES:10mM,EGTA:5mM,pH7.2。在实验期间,细胞和溶液在室温(19℃-30℃)保持。为了用手操作膜片钳记录,将细胞培养皿放置在显微镜的盘架上,并连续地用槽液灌流(1ml/min)。在膜片电极和细胞(吸液管电阻范围:2.5MΩ-6.0MΩ;封接电阻范围:>1GΩ)之间形成千兆欧姆的封接之后,将跨越吸液管尖端的细胞膜破裂,以使电进入到细胞内部(全细胞膜片结构)。对于使用QPatch系统的实验,将细胞以悬浮液形式转移到QPatch系统的槽液中,并进行自动化的全细胞记录。在-80mV的钳制电压下,对细胞进行电压钳。为了分析试验品,顺序预先培养浓度递增的试验品之后,用2μMGABA刺激GABA受体。预培养时间是30s,GABA刺激的时间是2s。将试验品溶于DMSO中,形成储备溶液(10mM)。在槽液中,将试验品稀释到0.01、0.1、1和10μM。在每种细胞上检验所有浓度的试验品。相对百分数增强定义为:在试验品存在下对GABAEC20响应的峰值,除以对单独的GABAEC20响应的峰值,乘以100。大鼠中的翻正反射丧失根据以下程序在雄性SpragueDawley大鼠中获得血浆药动学和镇静的定性评估。通过以范围为合适的媒介物中的5至15mg/kg的剂量经由足背静脉的静脉内推注剂量(60秒)施用于大鼠。为了评估镇静,用手将大鼠轻轻地约束到侧向位置用于剂量施用。如果在剂量施用期间观察到肌肉紧张度减少,则约束逐渐降低。如果动物不能返回到直立位置,则将时间记录为翻正反射丧失(LRR)的开始。在施用期间未发生LRR的情况下,此后通过置于背卧位以5分钟间隔评价动物。在30秒间隔内连续两次缓慢或不完全翻正符合翻正反射丧失。在LRR发作后,以相同的方式每5分钟评价动物。翻正反射的恢复定义为大鼠在被置于背卧位的20秒内完全自身翻正的能力。LRR的持续时间定义为LRR和翻正反射恢复之间的时间间隔。短期PTZ方法在与Giardina&Gasior(2009)CurrProtocPharmacol.,第5章中描述的方法类似的小鼠中戊四氮(pentylenetetazol)诱发的发作测定法中评估测试化合物的抗惊厥作用。在受控条件(温度22±2℃和12:12光照-黑暗循环,在8:00am光照)下在5组中圈养雄性CD-1小鼠,并且水和食物可随意获得。在行为测试之前将小鼠圈养1周,此时它们重25-35g。将戊四氮(PTZ,Sigma)以12mg/mL浓度溶于无菌的0.9%盐水中用于皮下施用。配制测试化合物,并在PTZ注射之前在预定时间点(通常30或60分钟)经由管饲(oralgavage)或腹膜内注射施用。将所有溶液制成新鲜的,并以10ml/kg体重的体积给予。在施用化合物之前使小鼠适应测试室至少30分钟。将小鼠随机分成至少四个测试组(媒介物和至少三个剂量的测试化合物),每组10只小鼠。在化合物施用后,观察小鼠在预定时间点(30或60分钟)的镇静的定性评估。在药物预处理时间之后,给小鼠s.c.注射PTZ(120mg/kg)。在PTZ注射后立即将小鼠分别置于观察室(25x15x15cm)中,并开始三通道计时器。连续观察每只小鼠30分钟,并且由对处理不知情的观察者记录以下行为:1)持续3秒的阵挛性惊厥前潜伏期,然后没有翻正反射,2)强直性惊厥前潜伏期,其特征在于所有四肢的刚性延伸,其超过与身体的90度角,3)死亡前潜伏期,4)阵挛性和强直性痉挛的数量。数据表示为平均值±S.E.M,并使用Dunnett或Bonferroni的事后检验的单因素方差分析来检测媒介物和剂量组之间的潜伏期和数量的显著差异。认为p值<0.05是统计学显著的。表1:例示性化合物的TBPS结合。对于表1,“A”指1nM至50nM的IC50,“B”指示IC50>50nM至100nM,“C”指IC50>100nM至500nM,并且“D”指示IC50>500nM。表2:在GABAA-R处的示例性化合物的电生理评估。对于表2:GABAA受体α1β2γ2和α4β3δ%效力:“A”10-100,“B”>100-500,“C”>500;D指示数据不可用或者尚未侧而定。*在克列莫佛中表3:翻正反射丧失(RatIV,5mpk)化合物大鼠LRR的持续时间SV-2BSA-3BSE-4ASA-7BSB-5BSV-5BSF-3ASV-3ASI-4BA≤20min;B>20minLRR:翻正反射丧失表4:最小有效抗惊厥药剂量定义为在PTZ处理小鼠中显著降低强直发作前的潜伏期的最低剂量A≤1mpk;B>1-3mpk;C>3mpk;PO–口服施用;IP–腹膜内注射。其它实施方案在权利要求中,冠词例如“一个(种)(a,an)”和“所述(该)(the)”是指一个(种)或超过一个(种),除非相反地说明,或者以其它方式从上下文是明显的。如果组成员中的一个、一个以上或全部存在于、用于给定的产品或方法中,或者以其它方式与给定的产品或方法相关,则认为在组的一个或多个成员之间包括“或”的权利要求或说明书被满足,除非相反地说明,或者以其它方式从上下文是明显的。本发明包括其中组中的正好一个成员存在于、用于给定的产品或方法或者以其它方式与给定的产品或方法相关的实施方案。本发明包括其中组成员中的超过一个或者全部存在于、用于给定的产品或方法中或者以其它方式与给定的产品或方法相关的实施方案。而且,本发明包括其中来自于一个或多个所列权利要求的一个或多个限定、要素、条款和描述性术语引入另外的权利要求中的所有变型、组合和排列。例如,任何从属于另一权利要求的权利要求,可以改变为包括在从属于同一引用基础权利要求的任何其它权利要求中得到的一个或多个限定。当要素作为列表例如以Markush组形式提出时,还公开了所述要素的各个子组,且可从所述组中除去任何要素。应理解,通常,当本发明或本发明的方面被称为包括具体的要素和/或特征时,本发明或本发明的方面的一些实施方案由这样的要素和/或特征组成、或者基本上由这样的要素和/或特征组成。为了简明起见,在本文中未以这些言词具体阐述那些实施方案。还注意,术语“包括”和“包含”为开放式的,且允许包括额外的要素或步骤。当给出范围时,包括端点。而且,除非另外说明或者以其它方式从上下文和本领域技术人员的理解是明显的,否则,作为范围表述的数值,可以将在本发明的不同实施方案中的陈述范围内的任何具体值或子范围采取为所述范围的下限的单位的十分之一,除非上下文清楚地另外规定。本申请参考多篇授权的专利、公布的专利申请、期刊文章和其它出版物,将其全部都通过参考引入本文中。如果在任何所引入的参考文献与本说明书之间存在冲突,则应以本说明书为准。另外,落入现有技术内的本发明的任何一些实施方案,可以从任意一个或多个权利要求中明确地排除。由于这样的实施方案被认为是本领域技术人员已知的,即使在本文中未明确地阐述排除,它们也可被排除。本发明的任何一些实施方案可以由于任何原因(不论是否与现有技术的存在有关)从任何权利要求中排除。本领域技术人员仅使用常规实验将认识到或能够确定本文中描述的一些实施方案的许多等同物。本文中描述的本实施方案的范围不限于以上说明书,而是如所附权利要求中所阐述的。本领域普通技术人员将理解,在不背离下列权利要求中定义的本发明的精神和范围的情况下,可对本说明书进行各种变化和改变。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1