吡咯并三嗪酮衍生物的制作方法
本发明属于医药化工领域,具体而言是涉及吡咯并三嗪酮衍生物和它们用于药物的用途。
背景技术:
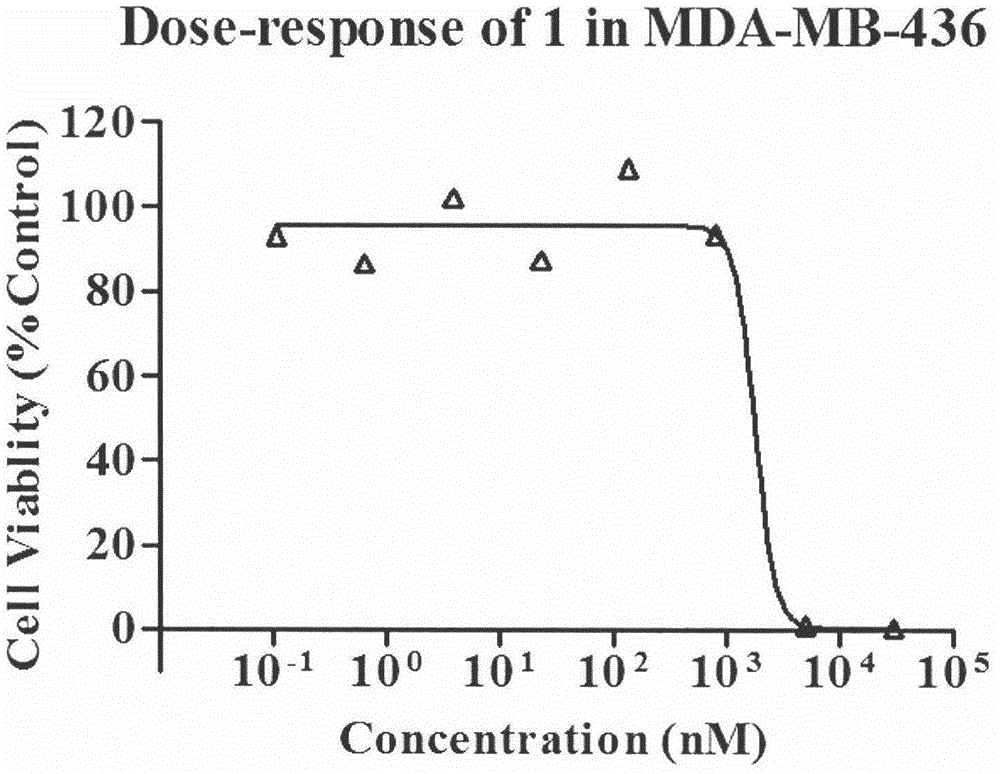
:本发明涉及的化合物有抑制聚(ADP-核糖)聚合酶活性的用途,该酶也称聚(ADP-核糖)合成酶和聚ADP-核糖基转移酶,普遍被称为PARP。该酶是存在于多数真核细胞内、具有多聚腺苷酸二磷酸核糖基(PAR)催化活性的蛋白酶,参与单链DNA损伤的感知与修复。该酶家族约有18种亚型,根据同源性程度的不同,将其分为3大组:PARP-1组,包括PARP-1~PARP-4,PARP-6,PARP-8和PARP-16;tankyrase组,包括PARP-5a~PARP-5c;第III组,包括PARP-7,PARP-9~PARP-15(NguewaPA,FuertesMA,ValladaresB,etal.ProgBiophysMolBiol,2005,88(1):143-172)。PARP-1是最先被发现的,在所有亚型中所占比例最大,发挥着90%以上的功能,包括介导DNA修复、通过消耗细胞能量池导致细胞机能障碍与坏死、促进炎症基因转录等,涉及到多种疾病的治疗,包括中风、神经退行性疾病、心肌缺血、癌症、炎症和糖尿病等(Peralta-LealA,Rodriguez-VargasJM,Aguilar-QuesadaR,etal.FreeRadicBiolMed,2009,47(1):13-26)。PARP-1主要由三个区域组成:(1)约46kDa的N端DNA结合区,包括细胞凋亡蛋白酶切割位点(caspase-3cleavagesite)、3个锌指结构以及核定位信号区(nuclearlocalizationsignal),这个区域通过ZnI、ZnII结构参与识别DNA的损伤以及介导PARP-1与DNA的结合,ZnIII则调节该区的活性;(2)一个22kDa的中心自修饰区域,包含15个高度保守的谷氨酸残基,被认为是自身聚腺苷二磷酸核糖化的靶位;(3)54kDa的C端催化区,包括形成烟酰胺腺嘌呤二核苷酸(NAD+)的结合位点和合成聚腺苷二磷酸核糖(PAR)的催化位点(LangelierMF,ServentKM,RogersEE,etal.JBioChem,2008,283(7):4105-4114)。PARP之所以成为肿瘤治疗中的热门位点之一,其理论依据是“合成致死”机制,即两个基因之间若存在“合成致死”作用,当其中任何一个基因单独受到抑制或发生突变时,细胞的生存不受影响,但同时抑制两个基因将导致细胞产生致死效应。乳腺癌、卵巢癌等患者的PARP与抑癌基因BRCA-1/2就是一对合成致死基因。抑制PARP的活性,可导致BRCA1/2突变的肿瘤细胞产生“合成致死”的效应。已有研究表明在DNA损伤后的同源重组修复中,缺乏一些较为关键的基因蛋白(包括BRCA1、BRCA2、XRCC1、XRCC2、RAD51、NBS1、FANCA、FANCC、CHK1、MRE11、RAD50、ATM、ATR等)的细胞对PARP抑制剂高度敏感,在PARP抑制剂的存在下无法对损伤的DNA片段进行有效的修复(KummarS,ChenA,ParchmentRE.etal.BMCMed,2012,10,25)。虽然根据很多研究表明PARP抑制剂单药对于同源重组修复(HR)缺陷的肿瘤细胞具有合成致死作用,如Bryant以及Farmer等的研究发现PARP抑制剂单药对于BRCA1和BRCA2突变的乳腺癌及卵巢癌细胞有明显的抑制作用(BryantHE,SchultzN,ThomasHD,etal.Nature,2005,434(7035):913-917;FarmerH,MccabeN,LordCJ,etal.Nature,2005,434(7035):917-921.),但是,更多的研究的重点则放在将PARP抑制剂与DNA损伤药物或放疗联合应用上,通过抑制由PARP介导的DNA的损伤修复,从而达到增强治疗效果,并通过减少用药(或放疗)剂量以达到降低毒副作用的效果(DamalG,DincalctIM.EurJCancer,2007,43(12):1791-1801;PeraltaLA,RodriguezMI,LinaresJL,etal.FreeRadicBiolMed,2009,47(1):13-26;ChalmersAJ.BrMedBull,2009,89(1):23-40;UnderhillC,ToulmondeM,BonnefolH.AnnOncol,2011,22(2):268-279)。奥拉帕尼(AZD-2281)是FDA批准的第一个PARP-1抑制剂,主要用于晚期卵巢癌患者。该药于2014年12月批准上市。本发明化合物可用于抑制PARP的活性。技术实现要素:本发明的第一方面提供式(1)的化合物:其中:X可以是NRX或CRXRY;如果X是NRX,则n是1或2,如果X是CRXRY,则n是1;RX可选自H,可被取代的C1-20烃基、C5-20芳基、C3-20杂环基、酰胺基、硫代酰胺基、酯、酰基以及磺酰基;RY取自H,羟基,氨基等;或者RX和RY可以一起构成螺-C3-7环烃基或杂环基;R2和R3都是H,或是当X=CRXRY时,R2,R3,RX和RY与它们所连接的碳原子一起可以构成可被取代的稠和芳香环;R1选自H或卤素;R4选自H或卤素。进一步的优选:下列优选能够应用于本发明的任一方面,只要适用的话。本发明中,吡咯可以被取代,但是在有些实施方案中优选为连接的原子本身连接于中心环羰基的邻位。因而,优选的取代位置如下式中的*所示:它通常被认为吡咯并三嗪酮部分的6-位,其取代的优选为连接的原子为H、Cl、F,更优选为F。R1优选为H、Cl、F,更优选为F。优选地,R2和R3都是氢。当n=2时,X是NRX,RX优选地选自下列基团:H;可被取代的C1-20烃基;可被取代的C5-20芳基;可被取代的C3-20杂环基;可被取代的酯基;可被取代的酰基;可被取代的酰胺基;可被取代的硫代酰胺基;和可被取代的磺酰基。当n=1时,X可以是NRX或CRXRY。在其中X是NRX的实施方式中,RX优选自下列基团:H;可被取代的C1-20烃基;可被取代的C5-20芳基;可被取代的C3-20杂环基;可被取代的酯基;可被取代的酰基;可被取代的酰胺基;可被取代的硫代酰胺基;和可被取代的磺酰基。在其中X是CRXRY的实施方式中,RY优选为氢。RX优选自下列基团:可被取代的C1-20烃基;可被取代的C5-20芳基;可被取代的C3-20杂环基;可被取代的酯基;可被取代的酰基;可被取代的酰胺基;可被取代的硫代酰胺基;和可被取代的磺酰基。本发明第二方面提供药物组合物,包含第一方面化合物和药学上可接受的载体或稀释剂。包括这些取代基熟知的离子、盐、溶剂合物和被保护形式。例如,对羧酸(-COOH)的称谓还包括阴离子(羧酸根)形式(-COO-)、其盐或溶剂合物以及其常规被保护形式。类似地,对氨基的称谓包括其质子化物(-N+HR1R2)、氨基的盐或溶剂合物以及氨基的常规保护形式。类似地,对羟基的称谓包括其阴离子形式(-O-)、其盐或溶剂形式以及羟基的常规被保护形式。异构体、盐、溶剂合物和被保护形式。某些化合物可以存在一种或多种特定的几何、旋光、对映、非对映、差向异构、立体异构、互变异构、构象或端基异构型,包括但不限于顺式(cis)与反式(trans)型;E-与Z-型;c-、t-与r-型;内-与外-型;R-、S-与内消旋-型;D-与L-型;d-与l-型;(+)与(-)型;酮基、烯醇与烯醇化物-型;顺(syn)-与反(anti)-型;顺错-与反错-型;α-与β-型;轴向-与平伏-型;船-、椅-、扭曲-、信封-与半椅-型;及其组合,一下统称为“异构体”(或“异构型”)。如果化合物是结晶的形式,则它可以存在多种不同的多晶型。注意的是除了下文关于互变异构体的讨论,特别要从本文所用的术语“异构体”中排除在外的是结构(或构造)异构体。例如对甲氧基-OCH3的称谓不被解释为对其结构异构体即羟甲基-CH2OH的称谓。但是,对一类结构的称谓也可以包括属于这种类别的结构异构体。例如,甲氧基苯基包括邻-、间-、和对-甲氧基苯基;丙基包括正丙基和异丙基。上述的排除不包括互变异构型,例如酮-、烯醇-和烯醇化物-形式。例如下列的互变异构对:酮/烯醇、亚胺/烯胺、酰胺/亚氨基醇、脒/脒、亚硝基/肟、硫酮/烯硫醇、N-亚硝基/羟基偶氮和硝基/酸式硝基。与本发明特别相关的是下列互变异构对:值得注意的是:在术语“异构体”中特别包括具有一个或多个同位素取代的化合物。例如,H可以是任意同位素形式,包括1H、2H(D)、3H(T)。类似地,C可以是任意同位素形式,包括12C、13C、14C;O包括16O、18O;等等。除非有另有指定,对特定化合物的称谓包括全部这样的异构型,包括其(完全或部分)外消旋和其它混合物。制备和分离这类异构体的方法是本领域中已知的,或者用已知方式调整本文所教导的方法或已知方法而容易获得的。除非有另有指定,对特定化合物的称谓也包括其离子、盐、溶剂合物和被保护的形式以及它的不同多晶型。本发明的第三方面提供用在人或动物体治疗方法中的第一方面的化合物。本发明的第四方面提供如本发明第一方面所定义的化合物在制备用于抑制PARP活性的药物中的用途。本发明的其他方面提供如本发明第一方面所定义的化合物在药物制备中的用途,该药物治疗:血管疾病;脓毒性休克;缺血损伤;神经毒性;出血性休克;病毒感染;或着因PARP活性抑制而改善的疾病。本发明的另一方面提供如本发明第一方面所定义的化合物在药物制备中的用途,该药物用于癌症疗法的辅助手段或者用于强化电离放射或化学治疗剂对肿瘤细胞的治疗。本发明的其他方面提供因PARP抑制而改善的疾病的治疗,包括对需要治疗的对象给与治疗有效量的如第一方面所定义的化合物,优选药物组合物的形式和癌症的治疗,包括对需要治疗的对象给予同时或先后与电离放射或化学治疗剂组合的治疗有效量的如第一方面所定义的化合物、优选药物组合物形式。在本发明的其他方面中,化合物可以用于制备用于治疗癌症的药物,该癌症是同源重组HR依赖性的DNADSB修复活性缺陷的,或者用于治疗癌症患者,该癌症是HR依赖性的DNADSB修复活性缺陷的,包括对所述患者给与治疗有效量的化合物。HR依赖性DNADSB修复途径经由同源机理修复DNA的双链断链(DSB),以生成连续的DNA螺旋(K.K.Khanna;S.P.Jackson.Nat.Genet.2001,27(3):247-254)。HR依赖性DNADSB修复途径的组分包括但不限于以下几种:ATM(NM_000051)、RAD51(NM_002875)、RAD51L1(NM_002877)、RAD51C(NM_002876)、RAD51L3(NM_002878)、DMC1(NM_007068)、XRCC2(NM_005431)、XRCC3(NM_005432)、RAD52(NM_002879)、RAD54L(NM_003579)、RAD54B(NM_012415)、BRCA1(NM_007295)、BRCA2(NM_000059)、RAD50(NM_005732)、MRE11A(NM_005590)和NBS1(NM_002485)。其它相关的参与HR依赖性DNADSB修复途径中的蛋白质包括如EMSY调节因子等(Hughes-DaviesL,HuntsmanD,RuasM,etal.Cell,2003,115:523-535)。HR的详细组分可见于相关文献中(WoodRD,MitchellM,SgourosJ,LindahlT.Science,2001,291:1284-1289)。HR依赖性DNADSB修复缺陷的癌症可以包含或者由一种或多种这样的癌细胞组成,它们相对于正常细胞而言通过该途径修复DNADSB的能力降低或者取消,即HR依赖性DNADSB修复途径的活性可能在一种或多种癌细胞中降低或者取消。HR依赖性DNADSB修复途径的一种或多种组分的活性可能在患有HR依赖性DNADSB修复缺的个体的一种或多种癌细胞中被消除。HR依赖性DNADSB修复途径的组分在本领域中被充分表征,例如参见文献(WoodRD,MitchellM,SgourosJ,LindahlT.Science,2001,291:1284-1289)所述以及包括上文索列举的组分。在有些优选的方案中,癌细胞可能具有BRCA1和/或BRCA2缺陷的表型,即BRCA1和/或BRCA2活性在癌细胞中被降低或消除。具有这种表型的癌细胞可能是BRCA1和/或BRCA2缺陷的,即BRCA1和/或BRCA2的表达/或活性可能在该癌细胞中被降低或消除,例如可以借助编码性核酸的突变或多态性,或者借助编码调节因子的基因的扩增、突变或多态性,例如编码BRCA2调节因子的EMSY基因(Hughes-DaviesL,HuntsmanD,RuasM,etal.Cell,2003,115:523-535)。BRCA1和BRCA2是已知的肿瘤抑制基因,它们的野生型等位基因经常在杂合载体肿瘤中丧失(JasinM.,Oncogene,2002,21(58):8981-8993;TuttA.,AshworthA.TrendsMolMed.,2002,8(12):571-576)。BRCA1和/或BRCA2突变与乳腺癌的关联在本领域中已经被充分认识(RadiceP.J.ExpClinCancerRes.,2002,21(3):9-12)。编码BRCA2结合因子的EMSY基因的扩增抑制也与乳腺癌和卵巢癌有关。BRCA1和/或BRCA2中突变的载体也处于卵巢、前列腺和胰腺癌症的升高风险中。在有些优选的实施方案中,个体就BRCA1和/或BRCA2或其调节剂的一种或多种变异,如突变和多态性而言是杂合的。BRCA1和BRCA2变异的检测在本领域是熟知的(NeuhausenS.L.,OstranderE.A.Genet.Test,1992,1:75-83;ChappnisP.O.,FoulkesW.D.CancerTreatRes.,2002,107:29-59;JanatovaM.Neoplasm,2003,50(4):246-250;JancarkovaN.CeskaGynekol.2003,6(1):11-16)。BRCA2结合因子EMSY扩增的测定描述可见于Hughes-Davies的文章中(Hughes-DaviesL,HuntsmanD,RuasM,etal.Cell,2003,115:523-535)。与癌症有关的突变和多态性可以通过检测变异核酸序列的存在而在核酸水平上检测,或者通过检测变异(即突变或等位基因变异)多肽的存在而在蛋白水平上检测。附图说明图1~图4显示的是使用Alarmblue法测定受试样品1~6及阳性对照AZD2281在MDA-MB-436肿瘤细胞中的药物浓度-细胞活力图,用来测定受试样品对肿瘤细胞的半抑制浓度,即IC50。具体实施方式本发明化合物可以这样合成:使用式1化合物(见下式):其中R1如前文所定义,与式2化合物(见下式)反应:其中n、R2、R3和X如前文所定义,反应可以这样进行:在偶联试剂系统存在下,例如O-苯并三氮唑-N,N,N′,N′-四甲基脲四氟硼酸酯(TBTU)、2-(7-偶氮苯并三氮唑)-四甲基脲六氟磷酸酯(HATU)、1-乙基-(3-二甲基氨基丙基)碳化二亚胺盐酸盐(EDCI)/1-羟基苯并三氮唑(HOBT),在碱(例如N,N-二异丙基乙胺(DIEA))存在下,在溶剂如N,N-二甲基甲酰胺(DMF)或二氯甲烷(DCM)中,在0℃至所用溶剂沸点的温度范围内。一般检测方法:使用Agilent1260型HPLC-MS(AgilentDorshell120EC-C18,2.7μm,4.6mm×50mm;AgilentQuadrupoleMS;VWD范围190-400nm)进行分析。流动相A(含0.1%甲酸的水)和B(乙腈)按梯度使用:5%B达1分钟,10分钟后升至100%B,保持5分钟,流速1ml/min,在254nm下检测。1H和/或13C-NMR利用BrukerAscend核磁共振谱仪分别在400MHz和75MHz下记录。化学位移按百万分之分数(ppm)、相对于内标四甲基硅烷(TMS)的δ报告。除非另有规定,全部样品均溶于DMSO-d6或CDCl3中测定。关键中间体的合成:2-氟-5-(4-氧代-3,4-二氢吡咯并[1,2-d][1,2,4]三嗪-1-基-甲基)苯甲酸(A)的合成将5-溴-2-氟苯甲腈(10.0g,50.00mmol),碘化亚铜(0.48g,2.52mmol),PdCl2(PPh3)2(1.75g,2.49mmol)加入到四氢呋喃(200ml)中,于室温下搅拌10分钟,其间抽真空-氩气置换操作,共需循环操作三次,每次3分钟。之后用注射器先后加入三乙胺(21ml,151.50mmol),三甲基硅乙炔(7.1ml,50.24mmol),反应液由深红色变为黄绿色,缓慢升温至60℃,反应液逐渐变成黑色,保持温度反应18小时。TLC检测(5%EA/PE展开剂,KMnO4显色)反应完全。停止加热,冷却至室温后,加入饱和氯化铵水溶液(400ml)淬灭反应,分液,水相用乙酸乙酯萃取(100ml×3),合并有机相,并用饱和氯化钠水溶液润洗1遍(300ml),无水硫酸钠干燥,旋干有机相,过柱(2%~5%EA/PE为洗脱剂),得2-氟-5-三甲基硅烷乙炔基苯甲腈油状液体9.96g。将四正丁基氟化铵(35.9g,137.30mmol)溶于四氢呋喃(200ml),在室温下,缓慢滴加入2-氟-5-三甲基硅烷乙炔基苯甲腈(9.96g,45.83mmol)的四氢呋喃溶液中,反应液由棕色变成黑色。TLC监测(5%EA/PE展开剂,KMnO4显色)待反应完全后,加入饱和氯化铵水溶液(400ml)淬灭反应,分液,水相用乙酸乙酯萃取(100ml×3),合并有机相,并用饱和氯化钠水溶液润洗1遍(300ml),无水硫酸钠干燥,旋干有机相,过柱(2%~5%EA/PE为洗脱剂),得5-乙炔基-2-氟苯甲腈黄色固体5.86g(两步总产率88.1%),m/z(M+1)+146。将对甲苯磺酰叠氮(6.0g,30.42mmol),5-乙炔基-2-氟苯甲腈(5.3g,36.51mmol),氯化亚铜(0.3g,3.04mmol)加入氯仿(60ml)中,于室温下搅拌10分钟,其间抽真空-氩气置换操作,共需循环操作三次,每次3分钟。之后用注射器先后加入三乙胺(5.10ml,36.51mmol)和吡咯(6.30ml,91.27mmol)。室温下搅拌,TLC监测(20%EA/PE作展开剂,KMnO4显色),待反应完全后,加入饱和氯化铵水溶液(100ml)淬灭反应,分液,水相用二氯甲烷萃取(50ml×3),合并有机相,并用饱和氯化钠水溶液(200ml)润洗1遍,无水硫酸钠干燥,旋干有机相,得N-[2-(3-氰基-4-氟苯基)-1-(1H-吡咯-2-基)亚乙基]-4-甲基苯磺酰胺(7.95g,产率68.53%)。无需纯化直接进行下一步的反应。将N-[2-(3-氰基-4-氟苯基)-1-(1H-吡咯-2-基)亚乙基]-4-甲基苯磺酰胺(7.0g,18.35mmol),肼基甲酸乙酯(2.8g,26.89mmol)加入叔丁醇(50ml)中,于室温下搅拌10分钟,其间抽真空-氩气置换操作,共需循环操作三次,每次3分钟,最后加入叔丁醇钾(6.0g,53.47mmol)。缓慢升温至80℃,TLC监测(30%EA/PE作展开剂,KMnO4显色)。待反应完全后,冷却至室温,加入水(100ml)淬灭反应,并用1NHCl调节溶液PH至7左右。用乙酸乙酯萃取(100ml×3),有机相用饱和氯化钠水溶液润(200ml)洗1遍,无水硫酸钠干燥,旋干有机相,过柱(10%~15%EA/PE为洗脱剂),得2-氟-5-(4-氧代-3,4-二氢吡咯并[1,2-d][1,2,4]三嗪-1-基-甲基)苯甲腈(4.21g,产率86.27%)。将2-氟-5-(4-氧代-3,4-二氢吡咯并[1,2-d][1,2,4]三嗪-1-基-甲基)苯甲腈(1.8g,6.71mmol),氢氧化钠(1.4g,35.00mmol)加入水(30ml)中,室温搅拌10分钟,缓慢升温至90℃。TLC监测(5%MeOH/DCM作展开剂),待反应完全后,冷却至室温。滴加1NHCl溶液调节溶液PH,有白色固体出,继续滴加1NHCl溶液直至再无固体析出。过滤出固体,滤饼用20%EA/PE溶液润洗一遍,真空干燥,得2-氟-5-(4-氧代-3,4-二氢吡咯并[1,2-d][1,2,4]三嗪-1-基-甲基)苯甲酸白色固体1.8g(产率:93.3%);1H-NMR(400M,DMSO-d6):13.20(s,1H),11.84(s,1H),7.79(dd,1H),7.55(m,1H),7.22(m,1H),6.23(m,1H),4.14(s,2H);m/z(M+1)+288。环戊基哌嗪-1-基甲酮(B)的合成将环戊酸(1.0g,8.76mmol)溶于10ml氯化亚砜中,加热回流2小时。旋干氯化亚砜,并用二氯甲烷带2次(10ml×2),用10ml二氯甲烷溶解。将N-BOC哌嗪(1.7g,9.13mmol)溶于10ml二氯甲烷中,缓慢滴加入上面溶液中,室温下搅拌2小时,旋干二氯甲烷,真空干燥,得4-环戊基基甲酰基哌嗪-1-甲酸叔丁酯(2.3g,产率93%)白色固体,无需进一步纯化即可直接使用。将4-环戊基-甲酰基哌嗪-1-甲酸叔丁酯(2.3g,8.14mmol)溶于20ml无水甲醇中,在剧烈搅拌下通入干燥HCl气体,持续通入1小时后,溶液中由大量白色固体析出,停止通气。再室温搅拌1小时后,旋干反应液。加入20ml饱和碳酸钠水溶液,用二氯甲烷萃取3遍(30ml×3),合并有机相,先后用大量饱和碳酸钠水溶液、氯化钠饱和水溶液润洗,有机相用无水Na2SO4干燥,旋干,真空干燥,得环戊基-哌嗪-1-基甲酮(1.41g,产率95%)无色油状液体,1H-NMR(400M,CDCl3):3.57(t,2H,J=4Hz),3.47(t,2H,J=4Hz),2.84-2.87(m,4H),2.21(s,2H),1.77-1.65(m,6H),1.58-1.48(m,2H);m/z(M+I)+183,无需进一步纯化即可直接使用。其它胺类化合物原料可以用类似的方法合成或者直接购买。实施例1:1-[3-(4-环戊基羰基-1-哌嗪-1-甲酰基)-苄基]-3H-吡咯并[1,2-d][1,2,4]三嗪-4-酮(1)的合成将3-(4-氧代-3,4-二氢吡咯并[1,2-d][1,2,4]三嗪-1-基-甲基)苯甲酸(2.0g,7.43mmol)、环戊基-哌嗪-1-基-甲酮(1.4g,7.68mmol)、O-苯并三氮唑-N,N,N′,N′-四甲基脲四氟硼酸酯(TBTU)(3.6g,11.21mmol)和N,N-二异丙基乙胺(2.9g,22.44mmol)加入到二甲基甲酰胺(20ml)中,搅拌3小时。加入水(100ml),在搅拌下,用1NHCl调节反应液PH,由大量固体析出,继续滴加盐酸直至无固体析出为止。过滤,滤饼先后用大量水,少量乙醇润洗,真空干燥,得到1-[3-(4-环戊基羰基-1-哌嗪-1-甲酰基)-苄基]-3H-吡咯并[1,2-d][1,2,4]三嗪-4-酮(1)白色固体(3.0g,93.0%),1H-NMR(400M,DMSO-d6):12.21(s,1H),7.75(d,1H),7.37~7.45(m,3H),7.27(d,1H),6.91(d,1H),6.80(t,1H),4.13(s,2H),3.27~3.54(m,8H),1.53~1.65(m,8H),1.53~1.65(m,8H),0.82~0.88(m,1H);13C-NMR(75M,DMSO-d6):144.96、142.13、137.47、130.48、128.95、127.57、125.70、125.50、117.50、115.46、109.64、41.10、38.14、30.13、26.03;m/z(M+1)+434.2。类似的,下列化合物可以通过相似的方法合成:实施例2:1-[3-(4-环丙基羰基-1-哌嗪-1-甲酰基)-4-氟苯基]-3H-吡咯并[1,2-d][1,2,4]三嗪-4-酮(4)的合成将2-氟-5-(4-氧代-3,4-二氢吡咯并[1,2-d][1,2,4]三嗪-1-基-甲基)苯甲酸(1.5g,5.22mmol)、环丙基-哌嗪-1-基-甲酮(0.48g,5.58mmol)、O-苯并三氮唑-N,N,N′,N′-四甲基脲四氟硼酸酯(TBTU)(2.5g,7.79mmol)和N,N-二异丙基乙胺(2.0g,15.50mmol)加入到二甲基甲酰胺(15ml)中,搅拌2~3小时。加入水(100ml),在搅拌下,用1NHCl调节反应液PH,由大量固体析出,继续滴加盐酸直至无固体析出为止。过滤,滤饼先后用大量水,少量乙醇润洗,真空干燥,得到1-[3-(4-环戊基羰基-1-哌嗪-1-甲酰基)-4-氟苯基]-3H-吡咯并[1,2-d][1,2,4]三嗪-4-酮(4)白色固体(2.3g,98.0%),1H-NMR(400M,DMSO-d6):12.20(s,1H),7.74(d,1H),7.42~7.58(m,2H),7.25(t,1H),6.91(d,1H),6.80(d,1H),4.13(s,2H),3.19~3.76(m,8H),1.99~2.04(m,1H),0.72~0.75(m,4H);13C-NMR(75M,DMSO-d6):171.74、164.48、144.89、144.87、134.60、132.40、129.65、125.83、117.17、116.18、116.40、115.72、109.57、36.52、10.83、7.59、0.57;m/z(M+1)+452.2。类似的,下列化合物可以通过相似的方法合成:实施例3:为了评估化合物的抑制作用,利用下面测定法测定IC50值。使用Alarmblue法测定受试样品及阳性对照AZD2281在MDA-MB-436肿瘤细胞中的IC50,每个化合物均设置9个浓度梯度(包括0浓度),2复孔。MDA436细胞在MEM完全培养基(含有10%的胎牛血清,100U·mL-1青霉素,100μg·mL-1链霉素)中,在37℃,5%CO2,95%空气培养条件下传2~3代。实验第0天(D0),将处于对数生长期细胞,用0.25%的胰蛋白酶消化,制备成单细胞悬液并计数,调整细胞浓度为1×104细胞/mL,每孔100μL加入96孔细胞培养板内,96孔板置于相应条件孵箱内培养过夜。阳性对照AZD2281配制:称取适量AZD2281,加入适量DMSO,涡旋振荡使药物完全溶解,分装后放入-80℃冰箱保存。受试品配制:称取适量受试品,加入适量DMSO,涡旋振荡使药物完全溶解,分装后放入-80℃冰箱保存。铺板次日(D1),每孔加入100μL含系列浓度受试化合物的完全培养基或对照的完全培养液;D4时,更换含有相应药物浓度的新鲜培养液。D7时,采用Alarmarblue方法检测各孔荧光强度,并计算IC50。IC50由以下公式计算:Y=Min+(Max-Min)/[1+10(LogIC50-X)*Slope]Min、Max和Slope分别表示最小值、最大值和斜率各化合物测定的IC50结果如下:化合物IC50/nM11808293803528483715302762169当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1