一种邻菲罗啉衍生物及其制备方法和应用与流程
本发明涉及有机合成技术领域,具体涉及一种邻菲罗啉衍生物及其制备方法和应用。
背景技术:
贵金属钯具有特殊的结构,对氢气和氧气有特殊的吸附能力,因此,在工业催化领域占有无法取代的地位。钯除了具有优良的催化活性之外,在很宽的温度范围内能保持化学惰性且具有熔点高、耐摩擦、耐腐蚀、延展性强、热电稳定性强等特性,被广泛应用于国民工业的各个领域。目前,我国钯资源储量有限,产量很低,远远不能满足现代化学工业的需要,大部分依赖进口,且价格昂贵。现有技术中,钯金属的制备与来源主要通过以下三个途径:(1)天然含钯矿产资源的利用;(2)从铜镍硫化矿副产品中对钯进行回收利用;(3)工业废气钯催化剂等含钯二次资源的回收利用。通过这三种途径获得的钯等金属资源有限,且随着钯资源损耗的增加,难以实现可持续利用。镍是银白色金属,具有磁性和良好的可塑性,延展性良好,中等硬度,有好的耐腐蚀性,主要用于合金及用作催化剂。近年来核电由于能量密度大、污染少,不会排放造成温室效应气体而得到蓬勃发展,然而核电运行当中会不可避免地产生乏燃料。乏燃料后处理所产生的高放废液(HLLW),是一种高酸性、高放射性和高毒性的混合溶液,钯的半衰期为6.5×106年,放射性周期长、危害大且含量高,如果能够进行实现回收利用,意义重大。
技术实现要素:
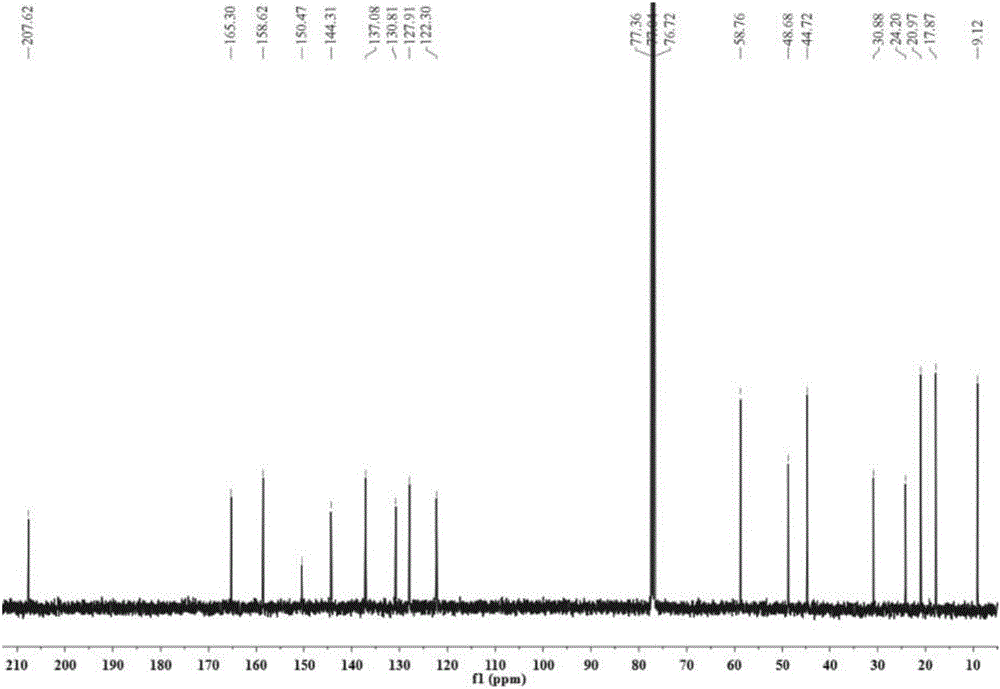
本发明提供了一种邻菲罗啉衍生物及其制备方法和应用,合成的邻菲罗啉衍生物与载体复合作为吸附剂,能够从酸性水相中吸附分离钯,选择性高,操作简单,分离效率高,适用于工业上分离回收贵金属钯。一种邻菲罗啉衍生物,如结构式Ⅱ所示:本发明还提供了一种所述的邻菲罗啉衍生物的合成方法,包括:步骤1,将如结构式Ⅲ所示的化合物Ⅲ溶于四氢呋喃中,得到混合溶液;步骤2,将混合溶液、如结构式Ⅳ所示的化合物Ⅳ、三乙胺混合后,搅拌回流反应直至原料反应完全,经后处理,得到如结构式II所示的化合物II。化合物II的名称为:2,9-二(5,9,9-三甲基-5,6,7,8-四氢-5,8-亚甲基苯并[e](1,2,4-三嗪-3-基))-2,2'-1,10邻啡罗啉,简称:CA-BOPhen。为了保证合成效率,优选地,化合物III和化合物Ⅳ的质量比为1:1~2。步骤2中的三乙胺作为催化剂,促进反应的进行,步骤2中,在搅拌状态下,将化合物Ⅳ和三乙胺(TEA)加入混合溶液中,搅拌回流反应至少7天,利用TLC跟踪反应直至原料耗尽后停止反应。为提高原料的利用率,降低后处理的复杂程度,优选地,化合物III与四氢呋喃的用量比为1g:50~60mL。化合物Ⅳ与三乙胺的用量比为1g:5~6mL。为了得到纯度较高的产物,优选地,步骤2中的后处理包括:依次对反应产物进行过滤旋蒸,以乙酸乙酯和石油醚的混合溶液作为展开剂,利用硅胶柱色谱分离技术进行分离纯化,纯化产物经真空干燥得到化合物II。作为优选,乙酸乙酯和石油醚的混合溶液中,乙酸乙酯和石油醚的体积比为2~3:1。作为优选,真空干燥的温度为80~90℃。本发明提供的化合物II对钯元素具有很好的吸附作用,本发明基于这种吸附作用,还提供了一种吸附分离钯的方法,包括如下步骤:将吸附剂与含有多种金属离子的硝酸水溶液混合,硝酸水溶液中的钯离子被吸附剂吸附分离,所述吸附剂由如结构式II所示的化合物负载在载体上制成。所述硝酸水溶液中含有Pd(Ⅱ)和其他金属离子,其他金属离子为Ni(Ⅱ)、Li(I)、Na(I)、K(I)、Rb(I)、Cs(I)、Ca(Ⅱ)、Mg(Ⅱ)、Sr(Ⅱ)、Ba(Ⅱ)、Nd(Ⅲ)、Co(Ⅱ)、La(Ⅲ)、Ru(Ⅲ)、Yb(Ⅲ)、Y(Ⅲ)、Fe(Ⅲ)、Zr(Ⅳ)、Mo(Ⅵ)中的至少一种。本发明合成的邻菲罗啉衍生物与载体复合作为吸附剂,能够从酸性水相中吸附分离钯,选择性高,操作简单,分离效率高,适用于工业上分离回收贵金属钯。附图说明图1为实施例1中化合物CA-BOPhen的核磁表征图谱;图2为实施例1中化合物CA-BOPhen的13CNMR谱图;图3为实施例1中化合物CA-BOPhen的MSI-MS表征图谱;图4为利用实施例4的吸附剂从硝酸水溶液中分离元素钯的分配系数随硝酸浓度变化的关系图;图5为利用实施例4的吸附剂从硝酸水溶液中分离元素钯的分配系数随接触时间变化的关系图。具体实施方式实施例1本实施例的合成路线如下所示:本实施例的合成步骤包括:(1)将4.5g樟脑醌(即化合物Ⅲ)置于500mL单口烧瓶中,加入250mLTHF将其溶解得到金黄色透明溶液;(2)在搅拌条件下向步骤1所得溶液中加入4.5g2.9-二甲酰胺腙-1,10-邻啡罗啉(即化合物Ⅳ)和24.0mL催化剂三乙胺(TEA)形成混合溶液,搅拌回流反应7天,利用TLC(薄层色谱分析)跟踪反应直至原料反应完全后停止。(3)反应停止后,冷却至常温抽滤出固体杂质,滤液经旋蒸出去溶剂得到油状物,采用乙酸乙酯/石油醚(体积比为2:1)作为展开剂,通过硅胶柱色谱分离技术进行分离纯化,纯化组分经80℃真空干燥后得到金黄色固体1.3g,即化合物Ⅱ(CA-BOPhen),产率20%。对化合物CA-BOPhen的结构进行表征如下:(a)1HNMR(400MHz,CDCl3,298K)在氘代氯仿中于25℃,测试了合成的CA-BOPhen化合物的1HNMR图谱,如图1所示,其中各峰归属为:δ(ppm):8.83(d,J=8.3Hz,2H),8.42(d,J=8.4Hz,2H),8.03(s,2H),3.54(d,J=3.6Hz,2H),2.04(d,J=5.1Hz,4H,加入重水后,峰消失为活泼H),1.74(s,4H),1.49(d,J=5.2Hz,4H),0.99(s,12H),0.83(s,6H)。图1中δ=7.2ppm处为氘代氯仿的质子峰,δ=5.3ppm处的单峰是溶剂二氯甲烷中质子形成的峰,化学位移和氢原子个数等指认结果均与CA-BOPhen的结构信息相符。(b)13CNMR(101MHz,CDCl3,298K)在氘代氯仿中于25℃,测试了合成的CA-BOPhen化合物的1HNMR图谱,如图2所示,其中各碳原子峰的归属为:δ(ppm):207.62,165.30,158.62,150.47,144.31,137.08,130.81,127.91,122.30,58.76,48.68,44.72,30.88,24.20,20.97,17.87,9.12。碳谱鉴定结果为该化合物中含有17个化学环境不同的碳,表明该化合物分子具有高度的对称性,结果与目标化合物结构相同,说明合成的化合物为目标化合物。(c)MSI-MS在甲醇溶液中测试了CA-BOPhen的MS-ESI阳离子图谱,结果见图3,分析结果表明:[M+H+]=591.6,[M+Na+]=613.5,计算值m/z为590.6。CA-BOPhen的理论分子量为590.6,此结果与CA-BOPhen分子量的理论值相符,MS-ESI结果分析说明成功合成了目标化合物。实施例2本实施例的合成步骤包括:(1)将4.5g樟脑醌(即化合物Ⅲ)置于500mL单口烧瓶中,加入225mLTHF将其溶解得到金黄色透明溶液;(2)在搅拌条件下向步骤1所得溶液中加入4.0g2.9-二甲酰胺腙-1,10-邻啡罗啉(即化合物Ⅳ)和20.0mL催化剂三乙胺(TEA)形成混合溶液,搅拌回流反应7天,利用TLC(薄层色谱分析)跟踪反应直至原料反应完全后停止。(3)反应停止后,冷却至常温抽滤出固体杂质,滤液经旋蒸出去溶剂得到油状物,采用乙酸乙酯/石油醚(体积比为1:1)作为展开剂,通过硅胶柱色谱分离技术进行分离纯化,纯化组分经80℃真空干燥后得到金黄色固体1.36g,即化合物Ⅱ(CA-BOPhen),产率20%。实施例3本实施例的合成步骤包括:(1)将4.5g樟脑醌(即化合物Ⅲ)置于500mL单口烧瓶中,加入270mLTHF将其溶解得到金黄色透明溶液;(2)在搅拌条件下向步骤1所得溶液中加入5.0g2.9-二甲酰胺腙-1,10-邻啡罗啉(即化合物Ⅳ)和24.0mL催化剂三乙胺(TEA)形成混合溶液,搅拌回流反应7天,利用TLC(薄层色谱分析)跟踪反应直至原料反应完全后停止。(3)反应停止后,冷却至常温抽滤出固体杂质,滤液经旋蒸出去溶剂得到油状物,采用乙酸乙酯/石油醚(体积比为3:1)作为展开剂,通过硅胶柱色谱分离技术进行分离纯化,纯化组分经80℃真空干燥后得到金黄色固体1.28g,即化合物Ⅱ(CA-BOPhen),产率20%。实施例4将0.5克实施例1制备得到的化合物CA-BOPhen溶解于70.0mL二氯甲烷中,充分溶解得到金黄色溶液;向此金黄色溶液中加入4.5克SiO2-P搅拌均匀,使SiO2-P与化合物混合均匀,经减压旋转蒸发使二氯甲烷挥发大部分至物料到近干状态,在毛细作用以及物理吸附作用下使有机分子进入SiO2-P孔径中,然后再将近干状态的物料在45℃下真空干燥24h,得到CA-BOPhen/SiO2-P复合材料,即吸附剂。SiO2-P是一种含多孔二氧化硅载体颗粒的有机高聚合物复杂载体,其制备方法如下:(1)将大孔的SiO2用浓硝酸洗涤、抽滤、去离子水洗至中性,重复10余次,干燥。(2)真空并有氩气保护条件下,以1,2,3-三氯丙烷和m-二甲苯为溶剂,向大孔SiO2中加入48.7g的m/p-甲酸基苯乙烯,8.9g的m/p-二乙烯基苯,72.2g二辛基临苯二甲酸酯,54.0g甲基安息香酸钠,0.56gα,α-偶二异丁腈和0.57g1,1′-偶二环己胺-1-腈,由室温逐步加热到90℃,并保持13小时,之后,逐步冷却至室温。(3)分别用丙酮和甲醇洗涤、抽滤上述产物,重复10余次,干燥。实施例5~11(1)将碱金属盐LiNO3、NaNO3、KNO3、RbNO3、CsNO3;碱土金属盐Mg(NO3)2、Ca(NO3)2、Sr(NO3)2、Ba(NO3)2;过渡金属盐Fe(NO3)3、(NH4)6Mo7O24·4H2O、ZrO(NO3)2、Co(NO3)2、Ni(NO3)2;贵金属Pd(5%w/w)硝酸盐溶液、Ru的硝酸盐溶液;稀土金属氧化物Y2O3、稀土金属硝酸盐La(NO3)3、Yb(NO3)3以及Nd(NO3)3等20种金属盐,溶于硝酸溶液加入去离子水配制成同时含有多种金属离子的硝酸水溶液,硝酸水溶液中的硝酸浓度为4.0M,各金属离子的浓度为2.0~5.0×10-3M。(2)在硝酸水溶液中加入浓硝酸和去离子水,调节硝酸水溶液中的硝酸浓度分别为0.4、1.0、2.0、3.0、4.0、5.0、6.0M,每种金属离子的浓度为5.0×10-4M。(3)将步骤(2)得到的含有20种金属元素的不同硝酸浓度的水溶液与实施例4制备的吸附剂接触混合,混合时的用量比为:每3.0mL硝酸水溶液对应0.15g吸附剂;(4)将步骤(3)所得混合液在TAITECMM-10型振荡器上进行吸附实验,振荡器振荡速率为150rpm,室温298K下操作,吸附时间为180min,使吸附达到平衡,然后测量吸附前后,不同硝酸水溶液中各金属元素的含量。实施例5~11的吸附结果如图4所示,图4中横坐标为硝酸浓度,纵坐标为分离系数。由图4可以看出,当硝酸浓度为0.4M时,分离元素钯的效果最好,钯的吸附率高于99%,且对于其他19种金属离子分配系数小于6,说明本发明提供的吸附剂对钯选择性强,适合用于从含有多金属元素水相中对钯进行高效分离回收,应用范围极广。实施例12~17实验条件以及步骤与实施例5相同,不同之处在于,将硝酸水溶液中,硝酸浓度固定为0.4M,依次改变接触时间为1、2、20、30、80、120min,所得分离结果如图5所示,图5中横坐标为硝酸浓度,纵坐标为分离系数。由图5可以看出,当接触时间为20min时,对钯的吸附已经达到了平衡状态,钯的吸附率高于99%,且对于其他19种金属离子分配系数小于6,说明本发明提供的吸附剂对钯选择性强,适合用于从含有多金属元素水相中对钯进行高效分离回收,应用范围极广。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1