一种特异靶向人ABCG2基因的sgRNA导向序列及其应用的制作方法
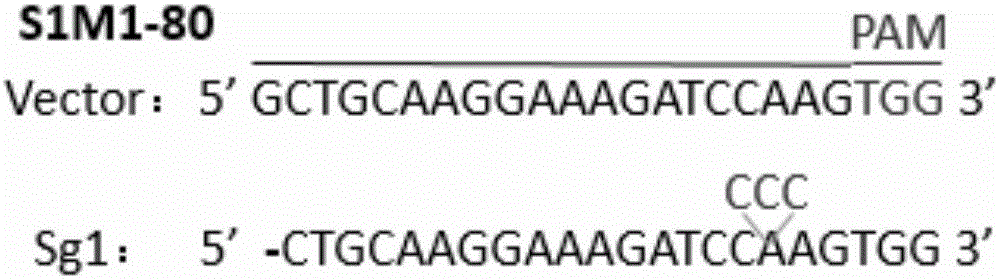
本发明属于基因工程应用领域,尤其涉及一种特异靶向人ABCG2基因的sgRNA导向序列及其应用。
背景技术:
:CRISPR-Cas9是细菌和古细菌在进化过程中演变出来的一种用于抵御外来侵害的免疫入侵系统,包括抵御入侵的病毒以及外源DNA。在现代基因工程应用领域与TALEN(transcriptionactivator-likeeffectornuclease)以及ZFN(zinc-fingernuclease)技术并列成为三大基因组编辑工具。相比较于TALEN及ZFN技术,CRISPR-Cas9技术具有特异性DNA识别能力,在第二类CRISPR系统中,Cas9核酸内切酶在sgRNA引导下切割双链DNA,造成基因组双链断裂,利用细胞基因组修复的不稳定性产生非特异重组来产生修复错误(插入或者缺失),从而可能产生移码突变而造成基因功能的丧失,实现基因敲除的目的。其sgRNA的设计和合成工作量远远小于TALEN和ZFN识别模块的构建过程,且毒性远远低于ZFN技术。但是CRISPR-Cas9技术也有上下文依赖性的缺点,目前只能应用于上游有PAM序列的靶位点。多药耐药性(MDR)是指对一种药物具有耐药性的同时,对其他结构不同,作用靶点不同的抗肿瘤药物也具有耐药性。多药耐药性是导致癌症药物治疗和肿瘤化疗失败的重要原因之一。而多药耐药性的主要机制之一就是ABC(ATP-bindingcassette)转运蛋白的过量表达,它们可由ATP水解供能来将抗癌药物泵出细胞外。它主要的家族成员包括ABCB1(P-GP)、ABCC1(MRP1)和ABCG2(BCRP)等。细胞中ABCG2的高表达将会导致多药耐药的发生从而使治疗失败,因此抑制或消除ABCG2的表达能够有效的解决在肿瘤治疗中出现的多药耐药问题。技术实现要素:为了克服某些高表达ABCG2蛋白的细胞出现多耐药的缺点与不足,本发明的首要目的在于提供一种特异靶向人ABCG2基因的sgRNA导向序列。该sgRNA导向序列可以用于敲除人ABCG2基因,进而抑制或消除ABCG2的表达。本发明的另一目的在于提供一种利用CRISPR-Cas9系统敲除人ABCG2基因的方法。该方法采用CRISPR-Cas9技术对ABCG2基因进行编辑,使其正常序列发生缺失或者突变从而达到敲除该基因的目的。本发明的再一目的在于提供上述特异靶向人ABCG2基因的sgRNA导向序列的应用。本发明的目的通过下述技术方案实现:一种特异靶向人ABCG2基因的sgRNA导向序列,为Sg1;其核苷酸序列为:Sg1:5'-GCTGCAAGGAAAGATCCAAG-3',位于基因ABCG2第三个外显子;一种利用CRISPR-Cas9系统敲除人ABCG2基因的方法,包含如下步骤:(1)在上述导向序列的5'端加上CACCG得到正向寡核苷酸;同时根据导向序列获得其对应的DNA互补链,并且在其5'端加上AAAC和其3'端加上C得到反向寡核苷酸;分别合成上述正向寡核苷酸和反向寡核苷酸,将合成的正向寡核苷酸和反向寡核苷酸变性,退火,形成双链;(2)将步骤(1)制得的双链与Cas9载体连接,得到重组敲除表达载体;(3)将步骤(2)制得的重组敲除表达载体与包装系统共转染包装细胞,然后收获病毒纯化并浓缩,得到病毒颗粒;(4)将步骤(3)制得的病毒颗粒感染细胞,筛选稳转细胞,得到成功敲除ABCG2基因的细胞。步骤(2)中所述的Cas9载体优选为lentiCRISPRv2载体;步骤(3)中所述的包装细胞优选为293T细胞;步骤(3)中所述的包装系统中的包装载体优选为pMD2.G和psPAX2;步骤(4)中所述的细胞优选为肿瘤多药耐药性细胞;步骤(4)中所述的细胞进一步优选为S1M1-80;所述的特异靶向人ABCG2基因的sgRNA导向序列在制备抗肿瘤多药耐药性药物中的应用;所述的肿瘤优选为人结直肠癌;本发明相对于现有技术,具有如下的优点及效果:(1)本发明根据sgRNA导向序列设计合成两条单链oligo序列,退火形成双链,然后与Cas9载体连接,利用Cas9载体将sgRNA以及CRISPR系统引入目标细胞中,Cas9蛋白会在sgRNA的引导下找到与其匹配的DNA序列,进行剪切,实现ABCG2基因的敲除。(2)载体中含有Puromycin抗性基因,利用Puromycin对细胞进行筛选,未转入lentiCRISPRv2载体的细胞将在筛选过程中被淘汰。(3)本发明提供的特异靶向人ABCG2基因的sgRNA导向序列,可通过CRISPR-Cas9系统敲除或编辑ABCG2基因,进而抑制或消除ABCG2的表达能够有效的解决在肿瘤治疗中出现的多药耐药问题。附图说明图1是特异靶向人ABCG2基因的sgRNA导向序列的序列及其位置示意图。图2是应用本发明设计的sgRNA对高表达ABCG2的细胞株进行编辑后抽提细胞mRNA逆转录成cDNA后PCR而后进行测序与成功转染空载lentiCRISPRv2的细胞株进行对比的测序分析图。图3是实施例2PCR产物测序结果的测序峰图。图4是成功敲除ABCG2基因的细胞株的ABCG2蛋白表达量的westernblot结果分析图。图5是Cisplatin、Doxorubicin和Mitoxantrone处理后,成功敲除ABCG2基因的细胞株的细胞活性结果分析图。图6是应用本发明设计的sgRNA对高表达ABCG2的细胞株进行编辑后的药物积累实验结果结果分析图。图7是实施例3药物积累实验的流式结果分析图。图8是实施例3药物积累实验流式结果的量化处理分析图。具体实施方式下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。实施例中所使用的细胞株均购自ATCC,lentiCRISPRv2载体购自Addgene,内切酶BsmBⅠ购自Biolabs,Polyetherimide(PEI)购自Ploysciences,Puromycin和Polybreen购自Sigma。实施例1(1)sgRNA设计根据人ABCG2基因的基因组序列(geneID:9429),设计1个靶向人ABCG2基因的sgRNA。20nt的寡核苷酸sgRNA导向序列为:Sg1:5'-GCTGCAAGGAAAGATCCAAG-3'(位于基因ABCG2第三个外显子)(图1);在其5'端加上CACCG得到正向寡核苷酸(Forwardoligo)(见表1中Sg1-F);根据导向序列获得其对应的DNA互补链,并且在其5'端加上AAAC和其3'端加上C得到反向寡核苷酸(Reverseoligo)(见表1中Sg1-R)。分别合成上述正向寡核苷酸和反向寡核苷酸,将合成的sgRNA寡聚核苷酸的Forwardoligo和Reverseoligo成对变性,退火;退火后可以形成连入表达载体lentiCRISPRv2载体的双链,同时将设计好的gRNA靶点序列进行blast比对以排除非特异性的靶切位点,具体寡核苷酸序列见表1。表1sgRNA导向序列的寡核苷酸序列Sequence(5'to3')Sg1-FCACCGGCTGCAAGGAAAGATCCAAGSg1-RAAACCTTGGATCTTTCCTTGCAGCC(2)构建表达sgRNA的载体病毒载体lentiCRISPRv2载体有BsmBⅠ酶切位点,用BsmBⅠ酶切,其中,酶切体系(20μL体系)为:BsmBⅠ1μL;10×NEbuffer2μL;质粒1μL;ddH2O16μL;酶切条件为:37℃酶切1h;将酶切后的载体lentiCRISPRv2分别与步骤(1)制得的退火双链利用T4连接酶进行连接,连接体系(10μL)为:退火双链(Sg1)2μL,lentiCRISPRv2载体2μL,10×NEBT4DNALigaseBuffer1μL,T4DNALigase1μL,ddH2O4μL;连接条件为:16℃连接过夜;将连接产物转化感受态细胞stbl3,具体转化方法为:-80℃取出感受态细胞stbl3,与冰上溶解;然后50μL感受态细胞中加入1μL的上述连接产物,混匀后冰浴30min;42℃水浴90s,过程中勿摇动;冰上冷却1~2min;然后加入800μLLB培养基,37℃摇床30min;涂AMP+板(100μg/mL)培养过夜,挑取阳性克隆后37℃摇床过夜进行扩大培养,并用HipurePlasmidMicroKitC(Magen)抽提质粒并测序验证,得到表达sgRNA的载体lentiCRISPRv2-hABCG2-Sg1(导向序列为Sg1)(hABCG2表示为人基因ABCG2)。实施例2(1)核心质粒与包装质粒pMD2.G和psPAX2共转染至293T细胞中培养293T细胞,待293T细胞的汇集率达到50%~60%,种板后12~18h为最佳转染时间;转染前更换新鲜培养液,60mm小皿中加入3mL培养基;转染时质粒的使用量为核心质粒(实施例1制得的lentiCRISPRv2-hABCG2-Sg1,另取lentiCRISPRv2空载体作为对照)4μg、psPAX23μg、pMD2.G1μg、PEI24μL,补充DMEM至总体积为200μL,加入顺序分别为DMEM、PEI和质粒DNA(核心质粒、pMD2.G和psPAX2);然后室温静止30min,使PEI与质粒DNA充分聚合,聚合结束后将转染体系逐滴加入上述培养有293T细胞的小皿中,轻轻摇匀后放入37℃、5%CO2培养箱中继续培养;(2)病毒的收获与浓缩收集转染后24h、48h、72h、96h293T细胞的上清液,每次收取病毒后再补充3mL新鲜培养液于小皿中,先收获的病毒原液用封口膜封闭后可暂存4℃冰箱中;待病毒液全部收集完成后1000rpm离心5min,以去除细胞碎片,用0.45μm滤膜过滤去除细胞碎片及其他杂质;取过滤后的病毒原液于100kD超滤柱内,4℃,4000g离心30min,收集滤膜内剩余约300μL病毒浓缩液,50μL每管分装于1.5mLEP管内,置-80℃可长期保存,避免反复冻融;(3)包装病毒颗粒分别感染目的细胞株S1M1-806孔板内提前一天种好待感染的目的细胞株S1M1-80(人结直肠癌多药耐药细胞),细胞感染时细胞汇集率达到40~50%为宜;感染前用1mL新鲜培养基换液,用另外1mL新鲜培养基稀释步骤(2)制得的50μL病毒浓缩液,加入2μL聚凝胺(Polybreen,10mg/mL,最终工作浓度为10μg/mL)混匀后,逐滴均匀滴入六孔板1个孔内,轻轻晃匀;依据不同细胞株的性质,于感染后6~48h换新鲜培养液;慢病毒介导的基因会在48~96h期间陆续表达,如果效率不理想可以进行反复感染,被感染表达目的基因的细胞理论上均为稳定株;核心载体有Puro(嘌呤霉素)的筛选标记,可以利用Puromycin进行稳定株的筛选;筛选之前测定实验所用细胞株Puromycin的全部致死浓度为30μg/mL,用全部致死浓度对感染后细胞S1M1-80进行筛选;(4)稳定感染细胞株的筛选与培养利用30μg/mL的Puromicin对感染后的细胞株S1M1-80进行筛选,筛选持续20d,对筛选后的细胞进行培养;(5)根据所设计的两段sgRNA序列设计鉴定引物,用于对敲除后目的片段进行鉴定,所设计引物如表2所示:表2鉴定引物序列Sequence(5'to3')Detection1-FGACCTGAAGGCATTTACTGAAGGAGCDetection1-RGCTCCATTCCGATCGAATCCCTTTTTCTTTC利用HiPureTotalRNAMiniKit对稳定感染细胞株S1M1-80进行mRNA提取,利用StarScriptⅡFirst-strandcDNASynthesisMix逆转录成cDNA,并利用鉴定引物进行PCR鉴定。其中,对含有lentiCRISPRv2-hABCG2-Sg1的病毒颗粒转染后的细胞株S1M1-80,其逆转录体系(20μL)为:RNA模板7μL;Oligo(dT)18(50μM)1μL;2×ReactionMix10μL;StarscripⅡRTMix1μL;DEPC-H2O2μL;逆转录条件:42℃孵育50min,85℃加热5min失活ScriptⅡ。对上述cDNA进行PCR,其反应体系(50μL):10×buffer5μL;dNTP1μL;模板DNA1μL;Detection1-F(10μM)1μL;Detection1-R(10μM)1μL;Pfu高保真酶1μL;ddH2O40μL;PCR反应扩增条件:95℃初始变性4min;95℃变性30S,56℃退火30S,72℃延伸2min,35个循环;72℃最后延伸10min;结果如图2和图3所示,表明:测序结果表明编辑组发生了缺失和插入突变,对照组没有发生变化。本发明发成功建立了敲除ABCG2基因的细胞株S1M1-80sg1。实施例3体外细胞实验对筛选后细胞进行鉴定人基因ABCG2敲除效果(1)westernblot利用westernblot实验鉴定实施例2中成功敲除ABCG2基因的细胞株S1M1-80sg1、的ABCG2蛋白表达量,成功转染空载体lentiCRISPRv2的细胞株S1M1-80作为空白对照,S1作为阳性对照,S1M1-80作为阴性对照;其中,一抗为Anti-ABCG2(MMSC-58222,购自santacruze),二抗为Anti-mouseIgG,HRP-linkedAntibody(货号7076,cellsignaling),具体方法为常规westernblot操作流程。结果如图4所示,在S1M1-80细胞中,转入sg1的细胞与只转入载体的细胞ABCG2表达量有明显差异,sg1中几乎未表达ABCG2蛋白,可作为ABCG2基因被敲除的证据之一。(2)MTT法检测细胞活性:选取ABCG2的特异性底物Mitoxantrone(购自LC)和Doxorubicin(购自LC)与非特异性底物cisplatin(购自LC)用MTT法检测检测成功敲除ABCG2基因的细胞株S1M1-80sg1对上述药物的敏感性。将细胞S1M1-80sg1及成功转染空载lentiCRISPRv2的细胞株S1M1-80对照细胞以每孔3000~5000个细胞的数量接种到96孔板,待细胞贴壁后,分别加入不同浓度的Mitoxantrone(1、3、10、30、100、300μM)、Doxorubicin(1、3、10、30、100μM)、cisplatin(1、3、10、30、100、300μM)。培养72h后,每孔加入10μL5mg/mL的MTT再培养4h,然后弃培养液每孔加入100μL的DMSO,用酶标仪在570nm读取每孔的吸光值。用Bliss法计算半数抑制浓度值(IC50)。结果如图5所示,与单独转入载体的对照细胞相比,成功敲除ABCG2基因的细胞株(S1M1-80)对Mitoxantrone和Doxorubicin的敏感性增加,IC50值明显降低。而对于ABCG2的非特异性底物Cisplatin来说,对照组与成功敲除ABCG2基因的细胞株对比,IC50值并没有差异。这可作为利用CRISPR-Cas9系统对S1M1-80的ABCG2基因进行敲除成功的证据之一。(3)药物积累实验基因ABCB2编码的高度糖基化跨膜蛋白ABCG2,它通过消耗ATP而将药物从细胞内泵出,从而降低肿瘤细胞内的药物浓度。选择3种ABCG2的底物:Mitoxantrone、Doxorubicin和Rh-123(购自sigma-Aldrich)流式观察这三种分子在细胞内的积累情况:将细胞S1M1-80sg1及成功转染空载lentiCRISPRv2的细胞株S1M1-80对照细胞以每孔2.5×105个的数量接种到6孔板,待细胞贴壁后,分别加入10μM的Mitoxantrone、Doxorubicin和Rh-123(Rhodamine123),培养2h后,PBS润洗3遍,然后将细胞用胰酶消化,用PBS重悬后用流式细胞分析仪检测荧光强度,然后用Flowjo软件进行定量分析;另外,选择3种ABCG2的底物:Mitoxantrone、Doxorubicin和Rh-123(购自sigma-Aldrich)共聚焦20倍镜观察这三种分子在细胞内的积累情况:先在6孔板里滴入少量培养基,然后放无菌盖玻片(22×22mm),将细胞S1M1-80sg1及成功转染空载lentiCRISPRv2的细胞株S1M1-80对照细胞以每孔2.5×105个的数量接种到6孔板,待细胞贴壁后,分别加入10μM的Mitoxantrone、Doxorubicin和Rh-123(Rhodamine123),培养2h后,吸掉培养基,用PBS清洗2次。用4%的多聚甲醛(溶剂为PBS)室温固定10min,吸掉多聚甲醛.加入PBS在摇床上快摇10min,然后加入新的PBS再快摇10min,如此操作2次,清洗掉残余的多聚甲醛。取200μLDapi溶液100nM对细胞进行染色,避光室温孵育15min。用PBS清洗盖玻片后置于载玻片(25×75mm)上,封片而后于共聚焦显微镜20倍镜下观察。结果如图6、7、8所示,编辑组(S1M1-80sg1)与对照组相比荧光强度明显增多,证明编辑组细胞的ABCG2没有发挥功能将三种药物泵出细胞。而对照组的ABCG2能正常的发挥药泵功能,因此荧光强度较低。表明编辑组ABCG2基因被成功敲除。上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 采用寡核苷酸介导的基因修复提高靶向基因修饰的效率的方法和组合物与流程
- CRISPR/Cas9靶向敲除人ezrin基因增强子关键区的方法及其特异性gRNA与流程
- CRISPR/Cas9 靶向敲除人PD‑1基因及其特异性gRNA的制作方法与工艺
- 靶向人TSPAN8基因的siRNA及其应用的制作方法与工艺
- 靶向人TSPAN8基因的siRNA及其应用的制作方法与工艺
- CRISPR/Cas9靶向敲除人乙肝病毒P基因及其特异性gRNA的制作方法与工艺
- 一种特异靶向人ABCG2基因的sgRNA导向序列及其应用的制作方法与工艺
- 一种利用CRISPR‑Cas9靶向敲除MSTN基因的方法与流程
- CRISPR/Cas9靶向敲除人CNE10基因及其特异性gRNA的制作方法与工艺
- 用于猪APN基因编辑的靶向sgRNA、修饰载体及其制备方法和应用与流程