用于制备前列腺素酰胺的新的方法与流程
技术领域
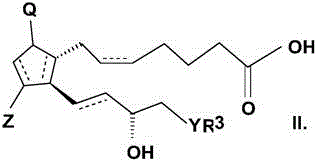
本发明的主题是用于制备通式(I)的前列腺素(prostaglandin)酰胺的方法。
在通式(I)的化合物中
取代基的含义如下:
用虚线标示的键可以是单键或双键,在位置5,6及13,14为双键的情形下,其可以是顺式或反式取向,
Q表示羟基且Z表示羟基或氧代基团,
R1及R2独立地表示氢原子或直链或支链的C1-10烷基或芳烷基,任选地经-ONO2、芳烷基或芳基取代,其含有杂原子,
R3表示直链或支链、饱和或不饱和的C4-6烃基,或C4-10烷基环烷基或环烷基,或任选地经烷基或卤素原子取代的苯基、C7-10烷基芳基或杂芳基,
Y表示(CH2)n或O原子或S原子,且
n=0-3。
背景技术:
为了经济地制备前列腺素酰胺衍生物,适当经取代的前列腺酸(prostan acid)须经活化。
根据目前的技艺状态,羧酸可以通过转化成其混合的酐、活化的酯或活化的酰胺而经活化,而且这些化合物可以随后通过与适当的胺反应而进一步转化成所要的前列腺素酰胺衍生物。
对于上述的可能性,化学性非常敏感的前列腺素酸通过酯形成的活化作用描述于例如EP 0 660 716中。
根据此方法,借助于烷基卤化物而形成起始的酯且该酯随后与适当的胺反应而得到酰胺官能团。此方法的缺点是要避免在合成结束时(即最后步骤)使用烷基卤化物,因为烷基卤化物经证明是具有遗传毒性的药物。此外,所得的酯必须在高温与适当的胺长时间反应且转化很少超过50%(EP0660716,42页,实施例12)。考虑前列腺素的已知的温度敏感性,其在高温的处理不利地影响以此方式所得的前列腺素衍生物的杂质分布及收率。
混合的酐的制备及其与经适当取代的胺的反应陈述于WO9153206中。
此方法的缺点是用于制备混合的酐的活性烷化剂-卤化的甲酸酯、特戊酰氯及其它-经证明是具有遗传毒性的化合物。
在WO2005058812(23页)陈述的方法中,起始的羧酸通过使用活化剂1-(3-二甲基氨基丙基)-3-碳二亚胺盐酸盐(EDC HCl)及乙胺而直接转化成乙基酰胺。在酰胺化反应期间,在位置11及15的羟基用四氢吡喃(THP)保护基保护,其随后被移除。
技术实现要素:
根据本发明,我们发现通过新的经活化的酯及新的经活化的酰胺,可以在温和的反应条件下,以高收率及高纯度制备通式(I)的化合物。
根据本发明,制备通式(I)的化合物可以通过使通式(II)的酸:
其中在式中
用虚线标示的键可以是单键或双键,在位置5,6及13,14为双键的情形下,其可以是顺式或反式取向,
Q表示羟基且Z表示羟基或氧代基团,
R3表示直链或支链、饱和或不饱和的C4-6烃基,或C4-10烷基环烷基或环烷基,或任选地经烷基或卤素原子取代的苯基、C7-10烷基芳基或杂芳基,
Y表示(CH2)n基团或O原子或S原子,且
n=0-3,
i)与合适引入R4的化合物反应,其中R4表示式a.)的基团,
且使得如此所得的通式(III)的酰胺:
其中Q、Z、R3、R4、Y及n的含义如上述定义,
与通式(IV)的胺反应
NHR1R2 IV.
其中R1及R2的含义如上述定义,或
ii)与合适引入R5的化合物反应,其中R5表示式b.)、c.)、d.)或e.)的基团,
其中(X)表示卤素原子或氢原子,
且使得如此所得的通式(V)的经活化的酯:
其中Q、Z、R3、R5、Y及n的含义如上述定义,
与通式(IV)的胺反应,其中R1及R2的含义如上述定义。
我们还发现根据本发明通式(I)的化合物也可以通过在2-氯-1,3-二甲基咪唑啉鎓氯化物及碱存在下,使通式(II)的化合物与通式(IV)的化合物反应而制备,其中在式中的取代基的含义如上述定义(方法iii)。
通式(III)及(V)的中间体是新的化合物。
对于合适引入R4的化合物,优选可使用1,1’-羰基二咪唑(DCI)或1,1’-硫羰基二咪唑,对于在存在活化剂的给定情况下引入R5的化合物,可使用N-羟基琥珀酰亚胺、N-羟基邻苯二甲酰亚胺、N-羟基-5-降冰片烯-内-2,3-二甲酰胺、1-羟基苯并三唑、(苯并三唑-1-基氧基)三(二甲基氨基)鏻六氟磷酸盐、N,N’-二琥珀酰亚胺基碳酸酯(DSC)或N,N’-二琥珀酰亚胺基草酸酯,特别是N,N’-二琥珀酰亚胺基碳酸酯。
对于活化剂,可使用N,N’-二异丙基碳二亚胺、N,N’-二环己基碳二亚胺或2-氯-1,3-二甲基咪唑啉鎓氯化物,优选N,N’-二异丙基碳二亚胺。
根据本发明在方法i)的过程中,可以在醚类溶剂或芳族溶剂或极性非质子溶剂或在它们的混合物中引入R4,使用例如二异丙醚、叔丁基甲基醚、2-甲基四氢呋喃、甲苯、茴香醚、二甲基甲酰胺、二甲亚砜、N-甲基吡咯烷酮,特别是四氢呋喃。所得的通式(III)经活化的酰胺在分离后或无分离时与通式(IV)的胺反应。引入基团R4期间的反应温度是在20-80℃之间,优选是70℃,而式(III)与(IV)化合物的反应是在20-80℃之间进行,优选在室温进行。
根据本发明在方法ii)的过程中,引入基团R5是在醚类溶剂或芳族溶剂或极性非质子溶剂或在它们的混合物中进行,使用例如二异丙醚、叔丁基甲基醚、2-甲基四氢呋喃、甲苯、茴香醚、二甲基甲酰胺、二甲亚砜、N-甲基吡咯烷酮,特别是四氢呋喃。所得的通式(V)经活化的酯在分离后或无分离时与通式(IV)的胺反应。引入基团R5期间的反应温度是在0-80℃之间,优选在室温,而式(V)与(IV)化合物的反应是在20-80℃之间进行,优选在室温进行。
根据本发明在方法iii)的过程中,反应是在醚类溶剂或芳族溶剂或极性非质子溶剂或在它们的混合物中进行,使用例如二异丙醚、叔丁基甲基醚、2-甲基四氢呋喃、甲苯、茴香醚、二甲基甲酰胺、二甲亚砜、N-甲基吡咯烷酮或四氢呋喃。至于碱,可以使用经常使用的碱,例如吡啶、N-甲基吗啉、二异丙基乙基胺、1,5-二氮杂二环[4,3,0]壬-5-烯、1,8-二氮杂二环[5,4,0]十一碳-7-烯或三乙胺。
反应是在0-70℃之间的温度进行,其通过向通式(II)化合物在有机溶剂中的溶液中在0-70℃优选30℃加入通式(IV)化合物、2-氯-1,3-二甲基咪唑啉鎓氯化物及2摩尔当量的碱。将混合物先在此温度搅拌且随后逐渐加热直到起始物质消失。通过TLC追踪反应。
方法i、ii或iii也可以在一锅法条件下进行。
对于通式(IV)的胺,在比马前列素的情形下,适于最终化合物的胺,可以使用乙胺。
对于通式(IA)化合物的制备
其中R1、R2、R3、Y及n的含义如上述定义,
使用根据本发明通式(IIA)的化合物作为起始物质。
通式(IIA)的化合物:
其中在式中
R3表示直链或支链、饱和或不饱和的C4-6烃基,或C4-10烷基环烷基或环烷基,或任选地经烷基或卤素原子取代的苯基、C7-10烷基芳基或杂芳基,
Y表示(CH2)n基团或O原子或S原子,且
n=0-3,
可以根据本发明制备,其通过将通式(XII)的内酯二醇(lactondiol):
其中R3、Y及n的含义如上述定义,
还原为通式(XIII)的内酯三醇(lactontriol):
其中R3、Y及n的含义如上述定义,
随后移除式(XIII)化合物的保护基,
且将如此得到通式(XIV)的化合物,
其中R3、Y及n的含义如上述定义,
通过Wittig反应而转化成通式(IIA)的化合物。
通式(XII)化合物的还原可以通过已知方法进行,例如用二异丁基氢化铝在四氢呋喃介质中。保护基可以通过已知方法在酸性或碱性介质中移除,优选在碱性介质中移除。
通式(XIII)的内酯三醇衍生物:
其中R3表示直链或支链、饱和或不饱和的C4-6烃基,或C4-10烷基环烷基或环烷基,或任选地经烷基或卤素原子取代的苯基、C7-10烷基芳基或杂芳基,
Y表示(CH2)n基团或O原子或S原子,且n=0-3,
是新的化合物。
根据本发明的另一个具体实施方案,具体的通式IIA化合物式(IIB)化合物也可以制备成结晶形式,其中R3表示苯基且Y表示-(CH2)-基团。
式(IIB)化合物的结晶形式是新的化合物。
式(IIB)化合物可以以结晶形式制备,其通过向含有式(IIB)化合物的混合物中加入酯类及醚类溶剂的混合物。
根据此方法,使用二甲醚、乙醚、二异丙醚优选乙醚及二异丙醚作为醚型溶剂,及使用乙酸乙酯、乙酸甲酯、乙酸异丙酯优选乙酸异丙酯作为酯型溶剂。结晶作用是在-30℃及30℃之间进行,优选在0-25℃之间进行。将如此所得的结晶的混悬液搅拌1-24小时,优选8小时,随后过滤并用醚类溶剂洗涤,优选用二异丙醚洗涤。过滤后的结晶在25-50℃之间真空干燥,优选在35-40℃真空干燥。
通式(II)及(XII)的化合物可以通过已知的方法制备,例如描述于US 5359095、WO 93/00329。
本发明方法的优点是所要的比马前列素最终产物(如果需要时)可以通过结晶的比马前列素酸合成。本发明方法的另一个优点是所要的最终产物是通过新的中间体、结晶、活化的酯或酰胺合成,其(如果需要时)可以分离且(如果需要时)可以通过结晶或色谱法纯化。由于施加羧酸活化剂(例如DSC、DCI),不需要对位置9、11及15的仲羟基进行保护,没有观察到平行的反应例如二聚物形成,不论是在活化的酯或活化的酰胺的情形还是在最终酰胺形成的条件下,且本发明经活化的羧酸衍生物容易以高收率及纯度分离。
令人惊奇地发现本发明结晶经活化的羧酸衍生物可以通过结晶法容易地纯化以消除杂质且也可以在温和的反应条件下且以高收率简易地转化成所要的酰胺最终产物。
熟知的是在活性药物成分(API)的情形中,杂质的量是一个关键问题,在比马前列素的情形中,每种未知杂质的量必须低于0.1%。为了保持此非常严格的限制,根据本发明的方法使用比马前列素酸的结晶作用及活性羧酸衍生物的结晶作用代替在WO09153206中所述的昂贵的制备性HPLC拆分。
本发明的另一个实施方案是制备式(IB)的比马前列素的高熔点结晶形式II的方法。
通过本发明的方法,可以制备化学与热力学稳定的且无其它结晶形式的高熔点结晶形式II的比马前列素。
下面的专利申请是涉及比马前列素产物的结晶作用:US 2005/0209337A1、WO 2009/153206A2、US 2009/0163596A1。
在US 2009/0163596A1的实施例30中,比马前列素的高熔点结晶形式(DSC峰读取值为79℃)(进一步:结晶形式II)通过其X-射线衍射、其在KBr片中的IR光谱及其DSC及TGA曲线进行表征。
WO 2009/153206描述了通过制备性HPLC、随后通过结晶法而纯化比马前列素产物。结晶作用是由乙腈溶剂或从作为溶解溶剂的乙腈和作为沉淀溶剂的TBME(叔丁基甲基醚)进行。根据该描述,通过此方法可以制备比马前列素的高熔点结晶形式(DSC峰读取值为79℃)。通过重复该方法,申请人没有成功得到比马前列素的高熔点结晶形式。
US 2009/0163596描述了比马前列素的结晶形式I及其制备。比马前列素的结晶形式I通过其熔点(62-64℃)、DSC、X-射线衍射及IR研究而表征。其详细描述结晶方法,例如溶解方法(在接近沸点温度的有机溶剂中或有机溶解溶剂与沉淀溶剂的混合物中)、冷却方法、从母液分离沉淀的结晶形式及干燥方法(在低温的真空下)。比马前列素的高熔点结晶形式(DSC峰读取值为79℃)无法通过在US 2009/0163596所述的方法制备。
通过本发明的方法,可以制备比马前列素的化学与热力学稳定的、高熔点的结晶形式II(=高熔点的结晶形式(DSC峰读取值为79℃),其不含结晶形式I。形式II通过其熔点(72-78℃)、DSC研究及IR与X-射线粉末衍射研究而表征。
本发明方法的本质是,由在后处理且蒸发后含有比马前列素的反应混合物,或由比马前列素的任何结晶或非结晶的形式或由它们的任何比例的混合物,通过从质子或醚类溶剂的结晶作用,可以制备热力学稳定的纯的形式II。结晶方法如下:将计算量的溶剂添加至油性或结晶的粗比马前列素中,然后干燥并定期暴露至机械效应。
根据上述,本发明涉及用于制备式(IB)的比马前列素的结晶形式II的方法,其特征是将计算量的醚类或质子溶剂添加至在后处理且蒸发后的含有比马前列素的反应混合物或比马前列素的任何结晶或非结晶的形式或它们的任何比例的混合物中,如果需要时将所得的混合物暴露至机械效应,然后干燥并均匀化。
在上述方法中所得的结晶形式II的熔点是在72-78℃之间,根据DSC研究的吸热峰是在73-79℃之间且熔化热高于75焦耳/克。
图6、8及4分别呈现在根据本发明方法所得的结晶形式II的DSC曲线、X-射线粉末衍射曲线及IR光谱。
图5、7及3分别呈现结晶形式I的DSC曲线、X-射线粉末衍射曲线及IR光谱。
在根据本发明的方法中,申请人使用计算的量、适宜是20-60重量%、优选是35重量%的质子溶剂,特别是醇类例如甲醇、乙醇和/或水。优选使用水作为质子溶剂。
至于机械效应,申请人施加搅拌或刮擦,或两者。添加的溶剂通过干燥除去。干燥在-60℃及70℃之间的温度在真空进行,尤其在35℃在真空进行。
至于醚类溶剂,申请人施加计算的量、优选是2000-8000重量%的二甲醚、乙醚、二异丙醚,优选乙醚。添加的溶剂通过干燥除去。干燥是在低温通过使得氮气经过而进行,优选在0-(-)50℃之间通过使得氮气经过而进行。
本发明还涉及以下方面:
项1.用于制备通式(I)的前列腺素酰胺的方法,
-其中在式中
用虚线标示的键可以是单键或双键,在位置5,6及13,14为双键的情形下,其可以是顺式或反式取向,
Q表示羟基且Z表示羟基或氧代基团,
R1及R2独立地表示氢原子或直链或支链的C1-10烷基或芳烷基,任选地经-ONO2、芳烷基或芳基取代,其含有杂原子,
R3表示直链或支链、饱和或不饱和的C4-6烃基,或C4-10烷基环烷基或环烷基,或任选地经烷基或卤素原子取代的苯基、任选地经烷基或卤素原子取代的C7-10烷基芳基或任选地经烷基或卤素原子取代的杂芳基,
Y表示(CH2)n基团或O原子或S原子,且其中n=0-3,
所述方法的特征在于:
使得通式(II)的酸:
其中在式中
用虚线标示的键可以是单键或双键,在位置5,6及13,14为双键的情形下,其可以是顺式或反式取向,
Q表示羟基且Z表示羟基或氧代基团,
R3表示直链或支链、饱和或不饱和的C4-6烃基,或C4-10烷基环烷基或环烷基,或任选地经烷基或卤素原子取代的苯基、任选地经烷基或卤素原子取代的C7-10烷基芳基或任选地经烷基或卤素原子取代的杂芳基,
Y表示(CH2)n基团或O原子或S原子,且其中n=0-3,
i)与合适引入基团R4的化合物反应,其中R4表示式a.)的基团,
且使得如此所得的通式(III)的酰胺
其中Q、Z、R3、R4、Y及n的含义如上述定义,
与通式(IV)的胺反应
NHR1R2 IV.
-其中R1及R2的含义如上述定义,或
ii)与合适引入基团R5的化合物反应,其中R5表示式b.)、c.)、d.)或e.)的基团,
其中(X)表示卤素原子或氢原子,
且使得如此所得的通式(V)的经活化的酯
其中Q、Z、R3、R5、Y及n的含义如上述定义,
与通式(IV)的胺反应,其中R1及R2的含义如上述定义,或
iii.)在2-氯-1,3-二甲基咪唑啉鎓氯化物及碱存在下,与通式(IV)的胺反应,其中R1及R2的含义如上述定义。
项2.通式(III)的化合物,
其中在式中
用虚线标示的键可以是单键或双键,在位置5,6及13,14的双键的情形下,其可以是顺式或反式取向,
Q表示羟基且Z表示羟基或氧代基团,
R3表示直链或支链、饱和或不饱和的C4-6烃基,或C4-10烷基环烷基或环烷基,或任选地经烷基或卤素原子取代的苯基、任选地经烷基或卤素原子取代的C7-10烷基芳基或任选地经烷基或卤素原子取代的杂芳基,
Y表示(CH2)n基团或O原子或S原子,且其中n=0-3,
且
R4表示式a.)的基团
项3.通式(V)的化合物,
-其中在式中
用虚线标示的键可以是单键或双键,在位置5,6及13,14为双键的情形下,其可以是顺式或反式取向,
Q表示羟基且Z表示羟基或氧代基团,
R3表示直链或支链、饱和或不饱和的C4-6烃基,或C4-10烷基环烷基或环烷基,或任选地经烷基或卤素原子取代的苯基、任选地经烷基或卤素原子取代的C7-10烷基芳基或任选地经烷基或卤素原子取代的杂芳基,
Y表示(CH2)n基团或O原子或S原子,且n=0-3,
且
R5表示式b.)、c.)、d.)或e.)的基团,
其中(X)表示卤素原子或氢原子。
项4.项1的方法i)的方法,其特征在于合适引入该基团R4的化合物是1,1’-羰基二咪唑或1,1’-硫羰基二咪唑。
项5.项1的方法ii)的方法,其特征在于对于在存在活性剂的给定情形下引入基团R5,使用N-羟基琥珀酰亚胺、N-羟基邻苯二甲酰亚胺、N-羟基-5-降冰片烯-内-2,3-二甲酰胺、1-羟基苯并三唑、(苯并三唑-1-基氧基)三(二甲基氨基)鏻六氟磷酸盐、N,N’-二琥珀酰亚胺基碳酸酯或N,N’-二琥珀酰亚胺基草酸酯。
项6.项5的方法,其特征在于所施用的活化剂是N,N’-二异丙基碳二亚胺、N,N’-二环己基碳二亚胺或2-氯-1,3-二甲基咪唑啉鎓氯化物。
项7.项1的方法,其特征在于该反应是在醚型、芳族或极性非质子溶剂或它们的混合物中进行。
项8.项1的方法,其特征在于引入基团R4是在20至80℃之间的温度进行,优选在70℃的温度进行。
项9.项1的方法i)的方法,其特征在于式(III)与式(IV)化合物的反应是在20至80℃之间的温度进行。
项10.项1的方法ii)的方法,其特征在于该反应是在醚型、芳族或极性非质子溶剂或它们的混合物中进行。
项11.项1的方法ii)的方法,其特征在于引入基团R5是在0至80℃之间的温度进行。
项12.项1的方法ii)的方法,其特征在于式(V)与式(IV)化合物的反应是在20至80℃之间的温度进行,优选在室温进行。
项13.项1的方法i)或ii)的方法,其特征在于式(III)或式(V)的活化的羧酸衍生物通过结晶和/或色谱法纯化。
项14.项1的方法iii)的方法,其特征在于该反应是在醚型、芳族或极性非质子溶剂或它们的混合物中进行。
项15.项7、10或14的方法,其特征在于使用四氢呋喃作为溶剂。
项16.项1的方法iii)的方法,其特征在于该反应是在0至70℃之间的温度进行。
项17.项1的方法iii)的方法,其特征在于使用的碱是N-甲基吗啉、N,N-二异丙基乙基胺、1,5-二氮杂二环[4,3,0]壬-5-烯或1,8-二氮杂二环[5,4,0]十一碳-7-烯。
项18.项1的方法,其特征在于所使用的式(IV)的碱是乙胺。
项19.项1的方法i)、ii)或iii)的方法,其特征在于该反应是在一锅法条件下进行。
项20.用于制备通式(IIA)化合物的方法,
其中在式中
R3表示直链或支链、饱和或不饱和的C4-6烃基,或C4-10烷基环烷基或环烷基,或任选地经烷基或卤素原子取代的苯基、任选地经烷基或卤素原子取代的C7-10烷基芳基或任选地经烷基或卤素原子取代的杂芳基,
Y表示(CH2)n基团或O原子或S原子,且n=0-3,
所述方法的特征是将通式(XII)的内酯二醇:
其中R3、Y及n的含义如上述定义,
还原成通式(XIII)的内酯三醇:
其中R3、Y及n的含义如上述定义,
移除通式(XIII)化合物的保护基,并将所得的通式(XIV)的化合物:
其中R3、Y及n的含义如上述定义,
通过Wittig反应而转化成通式(IIA)的化合物。
项21.通式(XIII)的内酯三醇,
其中R3表示直链或支链、饱和或不饱和的C4-6烃基,或C4-10烷基环烷基或环烷基,或任选地经烷基或卤素原子取代的苯基、任选地经烷基或卤素原子取代的C7-10烷基芳基或任选地经烷基或卤素原子取代的杂芳基,
Y表示(CH2)n基团或O原子或S原子,且n=0-3。
项22.结晶形式的式(IIB)化合物,
项23.用于制备结晶形式的式(IIB)化合物的方法,其特征在于向含有式(IIB)化合物的混合物中加入酯型与醚型溶剂的混合物。
项24.项23的方法,其特征在于使用甲醚、乙醚、二异丙醚优选乙醚及二异丙醚作为醚型溶剂。
项25.项23的方法,其特征在于使用乙酸乙酯、乙酸甲酯、乙酸异丙酯优选乙酸异丙酯作为酯型溶剂。
项26.项23的方法,其特征在于结晶是在(-)30℃及30℃之间的温度进行,优选在0至25℃之间的温度进行。
项27.项23的方法,其特征在于将晶体的混悬液搅拌1至24小时,优选8小时。
项28.项23的方法,其特征在于将晶体混悬液过滤并用醚型溶剂洗涤,优选用二异丙醚洗涤。
项29.项23的方法,其特征在于将该过滤的晶体在25至50℃之间真空干燥,优选在35至40℃之间真空干燥。
项30.用于制备式(IB)的比马前列素的结晶形式II的方法,
其特征在于:在后处理及蒸发后,于含有比马前列素的反应混合物或比马前列素的任何结晶或非结晶形式或它们的任何比例的混合物中,加入计算量的醚型或质子溶剂,如果需要时将其暴露至机械效应,且随后干燥并均匀化。
项31.经项30的方法制备的结晶形式II,其特征在于其熔点是在72至78℃之间。
项32.经项30的方法制备的结晶形式II,其特征在于根据DSC研究,吸热峰在72至79℃之间且其熔化热高于75焦耳/克。
项33.项30的方法,其特征在于使用20-60质量%的质子溶剂,优选是35质量%的质子溶剂。
项34.项30的方法,其特征在于使用醇和/或水作为质子溶剂。
项35.项34的方法,其特征在于使用甲醇或乙醇作为醇。
项36.项34的方法,其特征在于使用水作为质子溶剂。
项37.项30的方法,其特征在于使用2000-8000质量%的醚型溶剂。
项38.项30方法,其特征在于使用二甲醚、乙醚、二异丙醚作为醚型溶剂。
项39.项30的方法,其特征在于使用搅拌或刮擦或两者作为机械效应
项40.项30的方法,其特征在于添加的溶剂通过干燥除去。
项41.项33及40的方法,其特征在于干燥在(-)60℃及70℃之间的温度在真空下进行。
项42.项37及40的方法,其特征在于干燥在低温优选在0至(-)50℃之间通过使得氮气经过而进行。
产物的鉴定是借助于下面的分析仪器而进行:
NMR光谱通过Bruker-Avance III-500MHz仪器而记录,DSC曲线通过Mettler-Toledo DSC 1/700仪器而记录,IR光谱通过Perkin-Elmer Spektrum 400 FT-IR光谱仪而记录,MS光谱通过Shimadzu LC-MS-IT-TOF仪器而记录。熔点通过Büchi Melting Point B-545装置确定。
本发明的其它细节是在实施例中描述,但本发明不受限于这些实施例。
附图说明
图1.实施例1a的内酯三醇的IR光谱。
图2.实施例1/c1的比马前列素酸的IR光谱。
图3.实施例14/a的形式I的IR光谱。
图4.实施例10.)的形式II的IR光谱。
图5.实施例14/a的形式I的DSC曲线。
图6.实施例10的形式II的DSC曲线。
图7.实施例14/a的形式I的X-射线衍射曲线。
图8.实施例10的形式II的X-射线衍射曲线。
图9.实施例1/c2.)的比马前列素酸的X-射线衍射曲线。
图10.实施例1/c2.)的比马前列素酸的DSC曲线。
图11.实施例5.)的比马前列素的MS光谱(正离子化)。
图12.实施例5.)的比马前列素的MSMS光谱。
具体实施方式
1.制备起始物质
a.)制备[1,1’-联苯]-4-羧酸((3aR,4R,5R,6aS)-六氢-4-[(1E,3S)-3-羟基-5-苯基-1-戊烯-1-基]-2-羟基-2H-环戊二烯并[b]呋喃-5-基)酯(PPB-内酯三醇)
将55克的[1,1’-联苯]-4-羧酸((3aR,4R,5R,6aS)-六氢-4-[(1E,3S)-3-羟基-5-苯基-1-戊烯-1-基]-2-氧代-2H-环戊二烯并[b]呋喃-5-基)酯(PPB-内酯二醇)的内酯基
在1000毫升四氢呋喃(THF)溶剂中在(-)65-(-)85℃用422毫升二异丁基氢化铝的己烷溶液(DIBAL-H)还原。将反应混合物用NaHSO4溶液破坏,将水相用乙酸乙酯萃取,将有机相用NaHCO3溶液洗涤并在40-50℃将溶剂除去。将粗物质蒸发后得到46.2克油状物。
所得的PPB-内酯三醇的结构式:
在叔丁基甲基醚(TBME):己烷混合物中结晶后,从粗油状物得到41.6克白色结晶。
熔点:91.1-91.7℃
实施例1a的内酯三醇的IR光谱如图1所示。
13C及1H NMR数据:
*重叠的1H NMR信号(在括号中的数字是指信号基团在PMR光谱中的位置编号,方向:朝向减少的位移)
**13C NMR信号与DMSO溶剂的多重谱线重叠。
b.)制备(3aR,4R,5R,6aS)-六氢-4-[(1E,3S)-3-羟基-5-苯基-1-戊烯-1-基]-2H-环戊二烯并[b]呋喃-2,5-二醇(内酯三醇)
将46.2克的[1,1’-联苯]-4-羧酸((3aR,4R,5R,6aS)-六氢-4-[(1E,3S)-3-羟基-5-苯基-1-戊烯-1-基]-2-羟基-2H-环戊二烯并[b]呋喃-5-基)酯(PPB-内酯三醇)油状物溶解在230毫升甲醇中且在加入6.6克K2CO3后在35-45℃去酰基化。在(-)5-0℃用0.5M磷酸溶液将反应混合物的pH调整至7-8。将沉淀的结晶滤出并用甲醇:水混合物洗涤。将母液蒸发,用乙酸乙酯萃取,将有机相通过Na2SO4干燥,将干燥的物质滤出并将产物通过加入己烷而结晶。得到26克白色结晶的物质。
产物的结构式:
熔点:98-103℃
13C及1H NMR数据:
*重叠的1H NMR信号(在括号中的数字是指信号基团在PMR光谱中的位置编号,方向:朝向减少的位移)。
c.)制备7-[(1R,2R,3R,5S)-3,5-二羟基-2-[(1E,3S)-3-羟基-5-苯基-1-戊烯-1-基]-环戊基]-5-庚烯酸,(5Z)-(比马前列素酸):
c1.)将108克4-羧基丁基溴化鏻(KBFBr)溶解在800毫升THF中并将溶液冷却至0-(-)5℃。在此溶液中先加入91克叔丁醇钾(KOtBu),且搅拌并冷却至(-10)-(-15)℃后,加入25克内酯三醇在THF中的溶液。当达到预期的转化后,用水将反应混合物破坏,随后加入EtOAc。将水相用EtOAc洗涤。将水相用NaHSO4溶液酸化至pH=2并用EtOAc萃取。将合并的有机相用15%NaCl溶液洗涤,通过Na2SO4干燥,过滤并蒸发。将残留物从乙酸乙酯与二异丙醚的混合物结晶。将结晶滤出并洗涤,将滤液蒸发。将所得的黄色油状物在硅胶上通过色谱法(用二异丙醚-丙酮洗脱剂)纯化。得到25.5克油状物。
所得的比马前列素酸的IR光谱如图2所示。
c2.)将实施例1/c1.)所得的产物溶解在60毫升乙酸异丙酯中并在搅拌下在其中加入40毫升乙醚。将少量的比马前列素酸的晶种添加至反应混合物中。在搅拌下逐渐冷却至0℃并在其中加入约60毫升二异丙醚。将混悬液在此温度搅拌一夜后过滤并用二异丙醚洗涤且在真空干燥,得到20.4克晶状比马前列素酸。
所得的比马前列素酸的DSC曲线显示在图5且X-射线粉末衍射曲线是在图9。
产物的结构式:
熔点:63.0-65.5℃
13C及1H NMR数据:
*部分或完全重叠的1H NMR信号(在括号中的数字是指信号基团在PMR光谱中的位置编号,方向:朝向减少的位移)。
2.)制备7-[3,5-二羟基-2-(3-羟基-5-苯基-戊-1-烯基)-环戊基]-5-庚烯酸(2,5-二氧代-吡咯烷-1-基)酯(活化的酯)
将27.5克实施例1/c2.)的比马前列素酸溶解在270毫升THF中并在室温在其中加入13.7克N,N’-二异丙基碳二亚胺,随后加入13.7克N-羟基琥珀酰亚胺。将混合物在此温度搅拌后倒在1N NaHSO4溶液及叔丁基甲基醚(TBME)的混合物上。分离各相。将有机相用1N NaHCO3溶液洗涤,将水性-碱性相用TBME萃取。将合并的有机相通过Na2SO4干燥,过滤并蒸发。将残留物从己烷:丙酮的混合物结晶,得到30.04克白色结晶状物质。
产物:
熔点:93.5-103.4℃
3.)将27.5克实施例1/c1.)的比马前列素酸溶解在270毫升THF中并在室温在其中加入11.5克碳酸钾及19.6克N,N’-二琥珀酰亚胺基碳酸酯。将反应混合物在搅拌下逐渐加热至60℃且随后倒在1N NaHSO4溶液及叔丁基甲基醚(TBME)的混合物上。分离各相。将有机相用1N NaHCO3溶液洗涤,将水性-碱性相用TBME萃取。将合并的有机相通过Na2SO4干燥,过滤并蒸发。将残留物从己烷:丙酮的混合物结晶,得到30.9克白色结晶状物质。
产物:
熔点:93.5-103.4℃
13C及1H NMR数据:
*重叠的13C NMR,DMSO信号。**,***,+,++,+++,#:重叠的1H NMR信号。
4.)制备7-[3,5-二羟基-2-(3-羟基-5-苯基-戊-1-烯基)-环戊基]-5-庚烯酸1,3-二氧代-1,3-二氢-异吲哚-2-基酯(活化的酯)
将2克的比马前列素酸溶解在20毫升THF中并在室温在此溶液中加入1克N-羟基-邻苯二甲酰亚胺和1毫升N,N’-二异丙基碳二亚胺。将反应混合物搅拌2小时后倒在1N NaHSO4溶液及叔丁基甲基醚(TBME)的混合物上。分离各相。将有机相用1N NaHCO3溶液洗涤,将水性-碱性相用TBME萃取。将合并的有机相通过Na2SO4干燥,过滤并蒸发。将残留物从己烷:丙酮的混合物结晶,得到1.5克白色结晶状物质。
产物:
熔点:83.2-84.5℃
13C及1H NMR数据:
$DMSO信号重叠的13C NMR。$$重叠的13C NMR信号。*,**,***,+,++:重叠的1H NMR信号。
5.)通过活化的酯制备7-[(1R,2R,3R,5S)-3,5-二羟基-2-[(1E,3S)-3-羟基-5-苯基-1-戊烯基]-环戊基]-N-乙基-5-庚烯酰胺,(5Z(-)比马前列素)
将27.5克的比马前列素酸溶解在270毫升THF中并在室温在此溶液中加入13.7克N,N’-二异丙基碳二亚胺,随后加入13.7克N-羟基琥珀酰亚胺。将混合物在室温搅拌。所得的活化酯不分离。
完成酯形成后,将70毫升乙胺在THF中的2M溶液添加至反应混合物中。将混合物搅拌直到达成预期的转化,然后将其倒在1N NaHSO4溶液及叔丁基甲基醚(TBME)的混合物上。分离各相。将有机相用1N NaHCO3溶液洗涤,将水性-碱性相用TBME萃取。将合并的有机相通过Na2SO4干燥,过滤并蒸发后得到25.4克油状物。
产物:
13C及1H NMR数据:
MS数据
MS光谱:如图11所示
预期的化学式:
C25H37NO4
测量的实际质量:438.2648[M+Na]+
预期的实际质量:438.2615[M+Na]+ΔM=3.3mDa及7.53ppm
C25H35NO3(M-H2O)
测量的实际质量:398.2655[M-H2O+H]+
预期的实际质量:398.2690[M-H2O+H]+ΔM=-3.5mDa及8.79ppm
MSMS(先驱离子:438.26):如图12所示
预期的化学式:
C25H35NO3(M-H2O)
测量的实际质量:420.2520[M-H2O+Na]+
预期的实际质量:420.2509[M-H2O+Na]+ΔM=1.1mDa及2.62ppm
C25H32NO3(M-H2O-5H)
测量的实际质量:394.2366[M-H2O-5H]+
预期的实际质量:394.2377[M-H2O-5H]+ΔM=-1.1mDa及2.79ppm
C25H30NO2(M-2xH2O-5H)
测量的实际质量:376.2258[M-2xH2O-5H]+
预期的实际质量:376.2271[M-2xH2O-5H]+ΔM=-1.3mDa及3.46ppm
6.)通过活化的酯制备比马前列素
将27.5克的比马前列素酸溶解在270毫升THF中并在室温在此溶液中加入11.5克碳酸钾及19.6克N,N’-二琥珀酰亚胺基碳酸酯。将反应混合物在搅拌下逐渐加热至60℃。所得的活化酯不分离。
活化的酯形成后,将70毫升乙胺在THF中的2M溶液添加至反应混合物中。当反应完成后将混合物倒在1N NaHSO4溶液及EtOAc的混合物上。将有机相用1N NaHCO3溶液洗涤,将水性-碱性相用EtOAc萃取。将合并的有机相用NaCl溶液洗涤并通过Na2SO4干燥。将干燥的物质滤出,将滤液蒸发后得到25.7克油状物。
产物:
7.)通过活化的酰胺制备比马前列素
将27.5克的比马前列素酸溶解在270毫升吡啶中并在其中加入13.7克1,1’-羰基二咪唑。将混合物在20-25℃搅拌直到发生活化的酰胺形成。所得的活化酰胺不分离。
将70毫升乙胺在THF中的2M溶液添加至在室温的反应混合物中并将混合物搅拌直到达成预期的转化。然后将混合物倒在1N NaHSO4溶液及叔丁基甲基醚(TBME)的混合物上。分离各相,将有机相用1N NaHCO3溶液洗涤并将水性-碱性相用TBME萃取。将合并的有机相通过Na2SO4干燥,过滤并将滤液蒸发后得到23.82克油状物。
产物:
8.)从纯化的经活化的酯制备比马前列素
将30.9克实施例3的经活化的酯溶解在270毫升THF中并在此溶液中加入70毫升乙胺溶解在THF中的2M溶液。反应完成后,将混合物倒在1N NaHSO4溶液及EtOAc的混合物上。将有机相用1N NaHCO3溶液洗涤。将水性-碱性相用EtOAc萃取。将合并的有机相用NaCl溶液洗涤并通过Na2SO4干燥。将干燥的物质滤出并将滤液蒸发。在所得的油状物中加入35质量%的水且将产物结晶。得到24.8克高于99.5%纯度的白色比马前列素结晶。
产物:
熔点:71.9-72.5℃
HPLC:99.6%比马前列素,低于0.3%反式-比马前列素,0.1%其它杂质
9.)根据方法iii.)制备比马前列素
将2.00克比马前列素酸溶解在20毫升四氢呋喃(THF)中并在30℃先加入1.29克2-氯-1,3-二甲基咪唑啉鎓氯化物及1.44毫升三乙胺,经10分钟搅拌后,加入2.57毫升乙胺在THF中的2M溶液。将反应混合物在1小时内逐渐加热至70℃并将混合物在此温度搅拌直到起始物质消失(约1小时)。通过TLC追踪反应。
反应完成后,将混合物倒在1N NaHSO4溶液及乙酸异丙酯(iPrOAc)的混合物上。将有机相用1N NaHCO3溶液洗涤,将水性-碱性相用iPrOAc萃取。将合并的有机相用NaCl溶液洗涤并通过Na2SO4干燥。将干燥的物质滤出并将滤液蒸发后得到1.41克油状物
产物:
10.)从粗比马前列素油状物制备比马前列素的结晶形式II
在根据实施例6制备的比马前列素油状物中,加入35质量%的纯水。将混合物充分搅拌后在最高35℃的温度真空干燥,且每2小时搅拌并刮擦。完成干燥后,将混合物均匀化。此产物的IR光谱显示在图4且此产物的DSC曲线显示在图6。生产的形式II的X-射线衍射曲线显示在图8。
收率:96.9%
熔点:78℃
DSC起点:73.56℃
11.)制备比马前列素的结晶形式II
将根据实施例5制备的粗比马前列素在加热下溶解在3000倍量的乙醚中。然后在(-)20-(-)30℃通过使得氮气缓慢通过将溶剂除去。将所得的结晶均匀化,或先暴露至机械效应且随后均匀化。
收率:94.4%
熔点:75.9℃
DSC起点:72.92℃
12.)制备比马前列素的结晶形式II
在根据实施例6制备的粗比马前列素中加入35质量%的甲醇。将混合物充分搅拌后在最高35℃的温度真空干燥,且每2小时搅拌并刮擦。完成干燥后,将混合物均匀化。
收率:95.8%
熔点:77.2℃
DSC起点:73.07℃
13.)制备比马前列素的结晶形式II
在根据实施例6制备的粗比马前列素中加入17.5质量%的纯水及17.5质量%的乙醇。将混合物充分搅拌后在最高35℃的温度真空干燥,且每2小时搅拌并刮擦。完成干燥后,将混合物均匀化。
收率:92.3%
熔点:72.9℃
DSC起点:72.96℃
14.)
a.)制备比马前列素的结晶形式I(根据专利申请US 20090163596的实施例38)
将5.2克的粗比马前列素从106克乙腈结晶:将混合物加热至接近沸点的温度,使热溶液冷却至室温并将混合物在此温度搅拌1小时,然后在0-5℃搅拌2小时。将沉淀的结晶滤出,用20克冷(0-5℃)乙腈洗涤并在0-5℃真空干燥1小时,在室温经半小时且在30-40℃经2小时。
得到4.3克的比马前列素的结晶形式I。其IR光谱显示在图3,其DSC曲线显示在图5且其形式I的X-射线衍射曲线显示在图7。
收率:83%
熔点:62.1℃
DSC起点:63.61℃
b.)从形式I开始制备比马前列素的结晶形式II
在根据实施例14a制备的比马前列素的结晶形式I中加入35质量%的纯水。将混合物充分搅拌后在最高35℃的温度真空干燥,且每2小时搅拌并刮擦。完成干燥后,将混合物均匀化。
收率:97.3%
熔点:77.7℃
DSC起点:73.14℃
15.)制备比马前列素结晶形式II及I的混合物的结晶形式II
在结晶形式II及I的50%-50%混合物中,加入35质量%的纯水。将混合物充分搅拌后在最高35℃的温度真空干燥,且每2小时搅拌并刮擦。完成干燥后,将混合物均匀化。
收率:97.6%
熔点:78.2℃
DSC起点:73.77℃。
- 还没有人留言评论。精彩留言会获得点赞!