表达外源蛋白的毕赤酵母、其构建方法及其诱导表达方法与流程
本发明涉及发酵工程和生物工程技术领域,具体涉及表达外源蛋白的毕赤酵母、其构建方法及其诱导表达方法。
背景技术:
毕赤酵母表达系统是上世纪80年代初期发展起来的一种新型的外源蛋白表达系统,相对于大肠杆菌表达系统而言,毕赤酵母表达系统具有明显的优越性,诸如:倍增时较短,发酵周期短;基因组简单,便于操作;培养条件相对简单,且易于进行高密度培养,进而可以获得较高的目的蛋白产量;对外源蛋白可以进行折叠、糖基化修饰,形成二硫键等;同时,它还避免了酿酒酵母分泌效率差、表达菌株不够稳定、表达质粒易丢失等缺陷;并且,就糖基化程度而言,毕赤酵母中加到外源蛋白每条侧链的平均长度为8-14个甘露糖残基,较之酿酒酵母每条侧链平均50-150甘露糖残基要短得多,不会产生过度糖基化现象;此外,毕赤酵母其自身分泌的胞外蛋白很少,这对于后期的蛋白纯化非常有利。
自20世纪80年代起建立的以毕赤酵母作为单细胞蛋白生产菌的高密度发酵方法已经成熟,干细胞产量可高达100g/L。种种优势使毕赤酵母成为近年来极受青睐的真核基因表达系统宿主。已用该系统成功表达了数百种来自病毒、细菌、真菌、动植物和人的蛋白,如人白介素、人血清白蛋白、肿瘤坏死因子、乙肝表面抗原、水蛭素衍生物等。
毕赤酵母是目前应用最广泛的外源蛋白真核表达系统之一,该系统基于一个高效的醇氧化酶1(alcohol oxidase 1,AOX1)启动子(AOX1 promoter,PAOX1),PAOX1的转录只响应甲醇的诱导,当以甲醇为唯一碳源时,PAOX1启动外源蛋白的表达;而当甘油存在时,抑制AOX1的表达。
技术实现要素:
针对上述问题,本发明提供了表达外源蛋白的毕赤酵母,其能解决现有技术中毕赤酵母在甘油诱导条件下AOX1表达量低的技术问题;此外,本发明还提供了该毕赤酵母的构建方法和诱导表达方法。
本发明提供一种突变的毕赤酵母GTP1基因,其特征在于,所述突变导致毕赤酵母不能编码甘油转运蛋白或编码的甘油转运蛋白没有活性,并能够在甘油存在条件下启动PAOX1的转录。所述GTP1基因序列如SEQ ID NO.1所示编码氨基酸序列如SEQ ID NO:2所示。
进一步地,所述突变为全部或部分序列缺失突变、插入突变、移码突变或点突变。
进一步地,所述的缺失突变为缺失GTP1基因的整个开放阅读框或者部分序列突变。所述的GTP1体外缺失片段序列如SEQ ID NO:3所示。
本发明还提供了包含所述突变基因的重组载体。包含重组载体的宿主细胞。
本发明还提供了一种表达外源蛋白的毕赤酵母菌株,其特征在于,所述毕赤酵母菌株含有权利要求1-5任意一项所述的突变的毕赤酵母GTP1基因。
进一步地,所述毕赤酵母菌株是由酵母菌株X-33突变GTP1基因所得。
进一步地,所述毕赤酵母菌株于2016年06月28日保藏于中国微生物菌种保藏管理委员会普通微生物中心,保藏号为CGMCC No.12718。
同时,本发明还提供了一种表达外源蛋白的毕赤酵母菌株表达系统,其特征在于,所述系统包括(1)权利要求1中所述的毕赤酵母菌株CGMCC No.12718,(2)含有启动子为PAOX1的外源蛋白表达载体;(3)碳源。
进一步地,所述毕赤酵母菌株的GTP1基因发生突变,GTP1基因序列如SEQ ID NO.1所示,使得突变后的毕赤酵母基因不能编码甘油转运蛋白或编码的甘油转运蛋白没有活性。
进一步地,所述碳源为甘油、甲醇或甘油与甲醇的混合物。诱导所述毕赤酵母表达外源蛋白的碳源为甘油,所述甘油添加后的体积浓度B2的范围为0<B2≤0.5%。诱导所述毕赤酵母表达外源蛋白的碳源为甲醇,所述甲醇添加后的体积浓度A1的范围为0<A1≤2%。
诱导所述毕赤酵母表达外源蛋白的碳源为甲醇与甘油的混合物,所述甲醇添加后的体积浓度A2的范围为0<A2≤2%,所述甘油添加后的体积浓度B1的范围为0<B1≤0.5%。进一步优选为,所述甲醇添加后的体积浓度A2为1%,所述甘油添加后的体积浓度B1为0.25%。
此外,本发明还提供了一种表达外源蛋白的毕赤酵母的诱导表达方法,其包括以下步骤,
步骤1,将含有启动子为PAOX1的外源蛋白表达载体转化至毕赤酵母中,所述毕赤酵母含有突变的毕赤酵母GTP1基因;
步骤2,将步骤1得到的毕赤酵母接种于培养基中培养;
步骤3,离心步骤2中获得的毕赤酵母菌,并重新培养至含有碳源的培养基中培养。
其中,所述GTP1基因序列如SEQ ID NO.1所示,编码氨基酸序列如SEQ ID NO:2所示。所述突变为全部或部分序列缺失突变、插入突变、移码突变或点突变。所述的缺失突变为缺失GTP1基因的整个开放阅读框或者部分序列突变。所述的GTP1体外缺失片段序列如SEQ ID NO:3所示。所述的毕赤酵母菌株于2016年06月28日保藏于中国微生物菌种保藏管理委员会普通微生物中心,保藏号为CGMCC No.12718。
其中,步骤2中所述培养基为YPD液体培养基,于30℃、230r/min培养至OD600为2~6。
其中,所述碳源为甘油、甲醇或甘油与甲醇的混合物。诱导所述毕赤酵母表达外源蛋白的碳源为甘油,所述甘油添加后的体积浓度B2的范围为0<B2≤0.5%。诱导所述毕赤酵母表达外源蛋白的碳源为甲醇,所述甲醇添加后的体积浓度A1的范围为0<A1≤2%。诱导所述毕赤酵母表达外源蛋白的碳源为甲醇与甘油的混合物,所述甲醇添加后的体积浓度A2的范围为0<A2≤2%,所述甘油添加后的体积浓度B1的范围为0<B1≤0.5%。优选为,所述甲醇添加后的体积浓度A2为1%,所述甘油添加后的体积浓度B1为0.25%。
其中,所述碳源的添加方式为,以体积浓度计,0~24h,向液体培养基中添加1%甘油;24h~48h,向液体培养基中添加1%甲醇;48h~72h,向液体培养基中添加1%甲醇和0.25%甘油;72h~96h,向液体培养基中添加1%甲醇和0.25%甘油。
此外,本发明还提供了表达外源蛋白的毕赤酵母的构建方法,其包括以下步骤:
步骤1,体外构建发生突变的GTP1基因片段,发生突变的GTP1基因片段不能编码甘油转运蛋白或编码的甘油转运蛋白没有活性;
步骤2,将步骤1中得到的发生突变的GTP1基因片段转化至酵母菌中;
步骤3,对步骤2获得的酵母菌进行筛选,获得GTP1基因发生突变的酵母株;
步骤4,对步骤3获得的酵母菌进行筛选,获得在甘油或甲醇、甘油混合条件下诱导具有AOX1酶活性的毕赤酵母株。
进一步地,所述毕赤酵母菌株的GTP1基因序列如SEQ ID NO.1所示,编码氨基酸序列如SEQ ID NO:2所示。所述毕赤酵母菌株的GTP1基因发生的突变为缺失突变、插入突变、移码突变或点突变。优选为,所述毕赤酵母菌株的GTP1基因发生的突变为缺失突变。
进一步地,所述的缺失突变为缺失GTP1基因的整个开放阅读框或部分序列,GTP1体外缺失片段序列如SEQ ID NO:3所示。
采用本发明后,通过对毕赤酵母的GTP1基因发生突变,使得突变后的毕赤酵母基因不能编码甘油转运蛋白或编码的甘油转运蛋白没有活性,并经实验证明经突变GTP1基因后的毕赤酵母能够在甘油或者甘油、甲醇混合培养基中诱导PAOX1的转录,进而使得外源蛋白得以表达。同时,采用本发明连续添加碳源的培养方式,使ΔGTP1-EGFP在最终收获时,细胞浓度高而且诱导表达的EGFP量高。
附图说明
图1-1为本发明的实施例中采用的载体pPICZB的基因结构图。
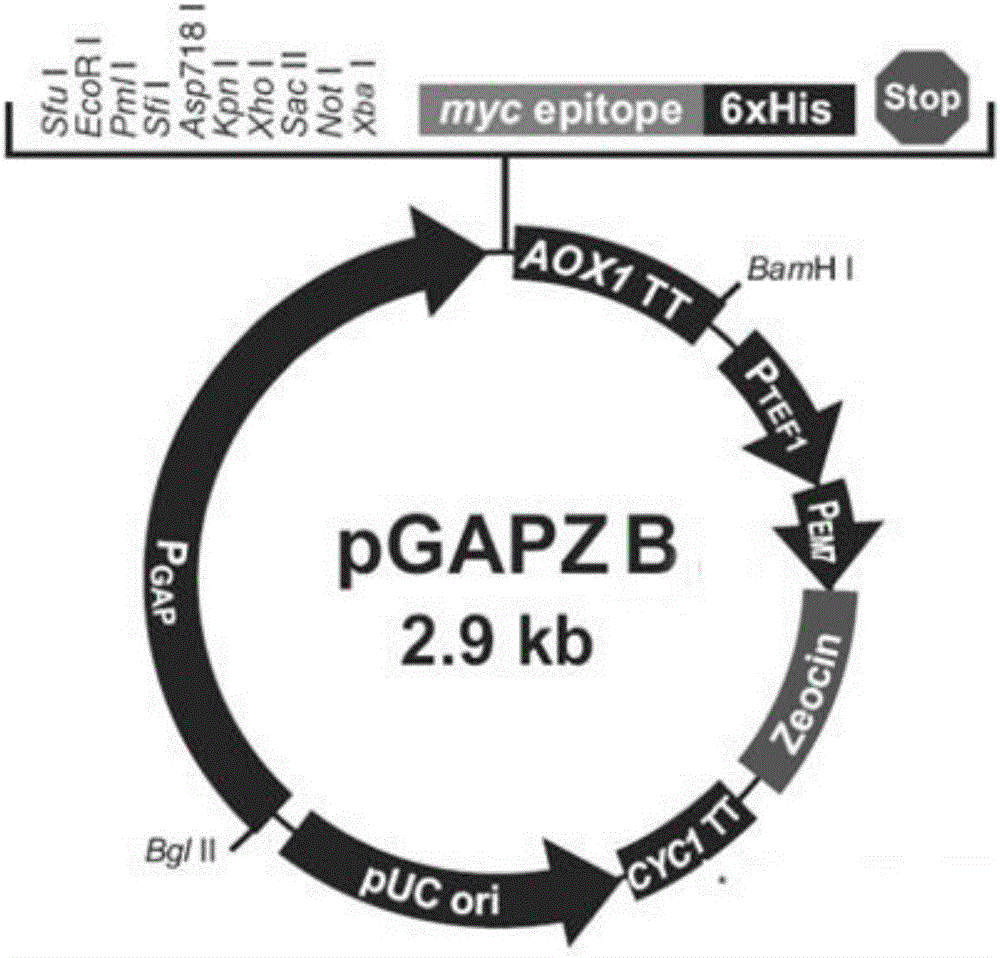
图1-2为本发明的实施例中采用的载体pGAPZB的基因结构图。
图2为本发明的实施例构建的ΔGTP1菌株和野生型菌株X-33分别采用不同碳源诱导AOX1酶活的测定结果图。图中1采用0.5%甲醇;2采用0.25%甘油;3采用0.5%甘油;4采用0.25%甘油和0.5%甲醇;5采用0.5%甘油和0.5%甲醇为碳源,上述甲醇、甘油均以添加后体积浓度计。
图3是本发明的实施例构建的表达菌株ΔGTP1-EGFP、x-EGFP和x-GGEFP分别采用不同碳源培养,分别对24h、48h、72h和96h荧光值的测定结果图。图中各字母代表如表1所示,表1中甲醇、甘油均以添加后的体积浓度计。
表1
图4是本发明的具体实施例构建的ΔGTP1-EGFP菌株和野生型菌株x-GGEFP在不同碳源诱导下的菌株生长曲线图,其中各字母代表的如表1所示。
图5是本发明的具体实施例构建的ΔGTP1菌株在优化培养条件下和在0.25%甘油和1%甲醇为碳源诱导下的菌株生长曲线图,其中,甲醇、甘油添加均以体积浓度计;优化培养条件如表2所示。
表2
图6是发明的具体实施例构建的ΔGTP1菌株在优化培养条件下和在0.25%甘油和1%甲醇为碳源下诱导EGFP表达的荧光值的测定结果图,其中,96h取样,甲醇、甘油添加均以体积浓度计。
图7是在整个发酵周期(0-96h),突变菌株ΔGTP1-EGFP在新的培养方式下与野生型x-GEFP在传统的培养方式下生物量(0D600)的比较。
具体实施方式
下面结合具体实施例,进一步阐述本发明。这些实施例仅用于说明本发明而不用于限定本发明的范围。
实施例1ΔGTP1菌株的构建方法
所用的试验材料及其来源包括:
DNA Marker、蛋白Marker、限制性内切酶(EcoRI、BamHI、HindIII、XbaI、KpnI、AvrII)、T4DNA连接酶、LA Taq DNA聚合酶,均购自TaKaRa公司。
质粒提取试剂盒,胶回收试剂盒,PCR产物柱纯化试剂盒,蛋白定量试剂盒,卡那霉素,博来霉素,G418抗生素购自上海生工生物工程公司。
引物由上海生工生物工程公司合成;酵母膏,蛋白胨(0X0ID,UK),其余试剂均为国产分析纯。
DNA电泳仪,凝胶成像仪,蛋白电泳仪(北京六一仪器),PCR仪(杭州朗基科学仪器有限公司),分光光度计(上海美诺达仪器有限公司),超声破碎仪(宁波新芝生物科技股份有限公司),酶标仪(Sunnyvale CA US Patent)等。
菌株、质粒和培养基:
克隆宿主菌E.coliDH5a,购自上海Sangon公司;毕赤酵母Pichia pastorisX-33(P.pastoris)、毕赤酵母过表达载体pGAPZB,pPICZB,购于Invitrogen公司,其中毕赤酵母Pichia pastoris X-33为野生型,以下简称为野生型X-33,大肠杆菌生长培养基LB和筛选培养基低盐LLB、毕赤酵母生长培养基YPD、诱导培养基BMMY、BMGY、BMMGY均由作者参考Invitrogen操作手册配制。
1.1GTP1体外缺失片段的构建
GTP1基因长1593bp(SEQ ID N0:1),编码530个氨基酸(SEQ ID NO:2),采用PCR,酶切连接的方法,以pMDTM19-T为载体在体外构建了GTP1体外缺失片段(SEQ ID NO:3),分别由GTP1基因上游片段410bp(SEQ ID N0:4)、G418抗性基因1464bp(SEQ ID N0:5)和GTP1基因下游片段363bp(SEQ ID N0:6)组成,所得GTP1体外缺失片段缺失了该基因的整个开放阅读框,避免了GTP1基因编码蛋白的产生。
具体操作如下:
a、以野生型X-33菌株基因组为模板,扩增GTP1基因上游片段;两段带有EcoRI和BamHI酶切位点,然后双酶切上游片段和质粒pMDTM19-T(TAKARA),T4连接酶16℃连接过夜,产生PGTPup质粒。
引物为
GTP1上游F:5‘-GCGAATTC CCGACAGAAGCAACCTCAGATCAACC-3’
GTP1上游R:5‘-AGGGATCC ATGGAGCGTPTAATCCGGAGTPGTPAAGAG-3′
b、以野生型X-33菌株基因组为模板,扩增GTP1基因下游片段,两段带有XbaI和HindIII酶切位点然后双酶切下游片段和PGTPup质粒,连接酶连接产生PGTP up-down质粒。
引物为
GTP1下游F:5‘-GCTCTAGA AACATCTCGTPTTCGTPGTPGCTTGTPGG-3‘
GTP1下游R:5‘-GCTAAGCTT CTTGCATTCGCTCAGGGCTCATTAC-3’
c、以pFA6a-KanMX6质粒为模板,扩增出G418抗性基因片段,两段带有BamHI和XbaI酶切位点,用这两种酶双酶切G418抗性基因片段和PGTP up-down质粒,连接产生敲除载体pMDTM19-T-GTP1-del。
引物为
kanF(G418):5‘-GCGGATCCCCGGTPTAATTAA-3’
kanR(G418):5‘-GCTCTAGAGAGCTCGTPTTAAAC-3’
1.2GTP1体外缺失片段电转野生型X-33菌株、筛选
电转:2.0Kv脉冲下,将GTP1缺失片段电转野生型X-33菌株。
筛选:将电转后的野生型X-33菌株涂于添加100ug/ml G418的YPD平板,放在30℃培养箱培养48小时。将平板上长出的单克隆挑至添加100ug/ml G418的YPD液体培养基中,30℃摇床培养后,提取基因组用PCR验证。把PCR检验及测序验证后正确的菌株命名为ΔGTP1。
实施例2AOX1酶活的测定
将保存于YPD平板的ΔGTP1菌株和野生型X-33接种于YPD液体培养基进行活化,选择对数期离心收集全部菌体,以体积浓度计,重悬于含有0.5%甲醇、0.5%甘油、0.25%甘油、0.5%甘油和0.5%甲醇,0.25%甘油和0.5%甲醇为碳源的YNB液体培养基中,培养48小时后提蛋白,经过Bradford法蛋白定量之后进行AOX1酶活得测定。
AOX1酶活得测定具体如下:在酶标板孔中加入100μL醇氧化酶检测溶液,然后加入提取的蛋白5μL,加入3μL甲醇启动反应,在室温反应15分钟-30分钟,直至显色,加入20μL2M硫酸(含0.1M亚硫酸钠)终止反应,并在492nm测量吸光度,图2为测定结果图,其具体数据如表3所示,ΔGTP1菌株可以在含有甘油和甘油甲醇培养基中诱导AOX1的表达,并且在甘油含量为0.25%时明显提高。
表3
实施例3诱导表达外源蛋白
3.1EGFP表达载体的构建
以EGFP为外源蛋白,如图1-1、图1-2所示,在载体pPICZB和pPGAPZB的A0X1启动子和GAP启动子的下游插入EGFP的编码序列,得到EGFP表达载体,分别为PA0X1-EGFP和Pgap-EGFP。
具体操作如下:用EcoRI和XhoI分别将质粒pMDTM19-T-EGFP、pPICZB、Pgapzb双酶切;pMDTM19-T-EGFP酶切产物EGFP用胶回收试剂盒回收纯化,酵母表达载体pPICZB和Pgapzb酶切产物用PCR纯化试剂盒回收;用T4连接酶连接片段和质粒,分别得到EGFP表达载体Paox1-EGFP和Pgap-EGFP,转化感受态E.coliDH5α,然后用含有博来霉素抗性的LLB平板筛选。
3.2表达菌株的筛选
2.0Kv脉冲下,分别将表达载体PAOX1-EGFP电转ΔGTP1菌株和野生型X-33菌株,涂于含有博来霉素抗性的YPD平板上,放在30℃培养箱48小时;将平板长出来的菌株挑至YPD液体培养基中,培养之后提基因组,用PCR验证正确的菌株命名为ΔGTP1-EGFP和x-EGFP;用同样的办法将表达载体Pgap-EGFP电转野生型X-33,最终将正确的菌株命名为x-GGEFP。
3.3拷贝数的确定
将上述构建好的3个表达菌株分别涂布于含有不同浓度的博来霉素(100μg/mL、200μg/mL、300μg/mL、500μg/mL、800μg/mL、1000μg/mL),最终挑取生长在800μg/mL而在1000μg/m生长很缓慢的平板上的三株菌株。
3.4多功能酶标仪检测EGFP表达量
挑取1.4.2获得的ΔGTP1-EGFP、x-EGFP和x-GGEFP单菌落,接种于装有YPD液体培养基的250mL的摇瓶,30℃、230r/min培养至OD600为2-6,4000r/min离心5min收集菌体,重悬于含有0.5%甲醇、0.5%甘油、0.5%甘油和0.5%甲醇,0.25%甘油和0.5%甲醇和0.25%甘油和1%甲醇为碳源的BMY液体培养基中,隔24h再次添加碳源,每次添加的碳源量相等,均为初次含有的碳源量,并取样品,0D定量后测荧光强度,激发波长为488nm,发射波长为505nm,其结果如表4和图3所示。
表4
从上表和图3中可以看出,野生型菌株只能在甲醇为碳源时,才能诱导AOX1高表达EGFP;菌株ΔGTP1-EGFP能在甘油培养基诱导AOX1表达EGFP,而野生型菌株低表达;在以0.25%甘油和0.5%甲醇为碳源的情况下,ΔGTP1-EGFP诱导AOX1表达的EGFP明显比野生型菌株高,尤其在48h时,高出将近6倍;ΔGTP1-EGFP在96时,其在以0.25%甘油和1%甲醇为碳源比0.25%甘油和0.5%甲醇为碳源诱导表达A0X1高出1.6倍;同时可以看出来诱导型启动子AOX1明显要比组成型启动子GAPDH就表达EGFP时要高出许多。
3.5表达菌株在不同碳源时的生长曲线
挑取1.4.2获得的ΔGTP1-EGFP、x-EGFP和x-GGEFP单菌落,接种于装有YPD培养基的250mL的摇瓶,30℃、230r/min培养至0D600为2-6,4000r/min离心5min收集菌体,然后转接到不同碳源的YNB和YP的液体培养基中,如表5所示,初始定量OD600为0.1,然后每隔8小时取样,测OD600值,其结果如图4所示。
表5
由图4可知,野生型菌株x-GGEFP在以0.5%甘油为碳源是最终0D可达到32.88,但是由图3可知表达的EGFP量较少;虽然ΔGTP1-EGFP在以1%甲醇为碳源是诱导AOX1表达的EGFP较高,但是其最终的0D为19.65相比在0.5%甘油为碳源的情况下0D差了13;ΔGTP1-EGFP在以0.25%甘油和1%甲醇为碳源时诱导表达EGFP量较高,而且最终0D为26.84。
实施例4优化ΔGTP1-EGFP的培养方式
由以上做的实验结果设计新的培养方式,使ΔGTP1-EGFP在最终收获时,细胞浓度高而且诱导表达的EGFP量高,因此设计了新的培养方式,如表2所示,以体积浓度计,0-24h,向液体培养基中添加1%甘油;24h-48h,向液体培养基中添加1%甲醇;48h-72h,向液体培养基中添加1%甲醇和0.25%甘油;72h-96h,向液体培养基中添加1%甲醇和0.25%甘油。同时,用传统的培养方式培养野生型菌株x-EGFP即:在前24h补加1%甘油,以后每隔24h补加一次1%甲醇诱导EGFP表达。
由图6可知,在96h时,单位OD600荧光值在以0.25%甘油和1%甲醇为10%,但如图5所示,最终细胞浓度优化培养时比以1%甲醇和0.25%甘油为碳源时要高出17%,即最终优化培养后的EGFP总的表达量比以1%甲醇和0.25%甘油为碳源时高出6.36%;
由图7可知,在整个发酵周期(0-96h),突变菌株ΔGTP1-EGFP在新的培养方式下比野生型x-GEFP在传统的培养方式下生物量(0D600)有显著差异,单位细胞浓度的荧光强度无显著性差异,最终优化培养后的EGFP总的表达量比野生菌在传统培养条件时高出23%。
以上实验证明本申请构建方法所获的表达外源蛋白的毕赤酵母能够在甘油或者甘油甲醇混合培养基中诱导PA0X1表达外源蛋白,并且本申请的上述优化后的诱导表达方法能够有效提高最终细胞浓度的。
需要说明的是,本发明中所提及的毕赤酵母全称为巴斯德毕赤酵母,该巴斯德毕赤酵母菌株于2016年06月28日保藏于中国微生物菌种保藏管理委员会普通微生物中心,保藏地址:北京市朝阳区北辰西路1号院3号,保藏号为CGMCC No.12718。
- 还没有人留言评论。精彩留言会获得点赞!