芳基腈类化合物的制备方法与流程
本发明涉及氰类化合物的合成领域,具体而言,涉及一种芳基腈类化合物的制备方法。
背景技术:
芳基腈类化合物不但是医药、农药、染料、功能材料、香料和天然产物等重要结构组成部分,而且也是有机合成重要的中间体。在一定的反应条件下芳基腈类化合物可以容易的转化为其他化合物如醛、酮、羧酸、胺、酰胺和杂环衍生物等。
Sandmeyer反应和Rosemund-Vonbraun反应是合成芳腈的经典方法,然而这两种方法通常需要化学计量的剧毒金属氰化物如氰化锌、氰化钠等,以及苛刻的反应条件(150~250℃)。过渡金属催化卤代芳烃的氰基化反应在最近十几年里得到了快速发展,但也存在一些问题:对空气和水敏感、催化剂价格昂贵且所需膦配体的合成复杂且收率低导致芳基腈类化合物合成成本较高,采用的氰源大多数是剧毒金属氰化物,限制了其在工业生产中的应用。
最近文献报道了使用低毒的黄血盐(亚铁氰化钾)为氰化试剂,钯或铜催化的芳环氰基化反应获得较为理想的收率(如申请号为200610048481.9的中国专利申请,Catal.Commun.2009.10 768–771,J.Am.Chem.Soc.2003,125,2890-2891,Eur.J.Org.Chem.2007,2401–2404,Tetrahedron Lett.2005,46 2585–2588);也有文献报道使用剧毒的金属氰化物为氰源,使用铜催化实现芳基的氰化反应,且这一类反应中均加入添加剂KI,但是反应温度仍然大于100℃,且收率中等。另外相关文献专利报道使用叠氮化钠在钯或铜催化剂条件下(申请号为200910088860和201510194625的中国专利申请)对各种取代芳基实现芳基氰基化,虽然这一类反应避免使用剧毒氰化物,但使用易爆炸的叠氮化钠和高成本的金属钯催化剂,不易于工艺规模化生产。
最近,有文献报道了芳基甲磺酸酯底物能在Ni或Pd催化剂条件下(J.Org.Chem.1995,60,6895-6903,SynLett.2014,25,2938–2942),以氰化钾为氰源实现中等收率的芳基氢化反应,但该类反应对底物的普适性较低,且底物甲磺酸酯合成条件苛刻、成本高、活性相对较低,需要在较高温度下实现。
技术实现要素:
本发明的主要目的在于提供一种芳基腈类化合物的制备方法,以解决现有技术中的芳基腈类化合物的制备成本较高的问题。
为了实现上述目的,根据本发明的一个方面,提供了一种芳基腈类化合物的制备方法,上述芳基腈类化合物具有通式I:制备方法包括:以具有通式II的芳基化合物为底物,其中,n=0~1,X1、X2、X3和X4中在化学可接受的结构中各自独立地选自N、S、O和C中的任意一种;Y为OSO2F、OTf或OTs;R1、R2、R3和R4各自独立地选自H、烷基、芳基和卤素中的任意一种,在催化剂、还原剂和配体的催化作用下使芳基化合物与氰源进行氰基化反应,得到芳基腈类化合物。
进一步地,上述氰源选自K4Fe(CN)6、Zn(CN)2、KCN和NaCN组成的组中的一种或两种。
进一步地,上述催化剂为非贵金属盐,优选非贵金属盐选自CuI、CuBr、CuCl、Cu(OAc)2、Cu(acac)2、Cu(OTf)2、CuI2、CuCl2、CuSO4、NiX2、Ni(OAc)2、NiX2(dppf)、NiX2(dppe)、NiX2(dppp)、Ni(PCy3)X2和Ni(Py)2Cl2组成的组中的任意一种或多种,其中X表示卤素。
进一步地,上述配体为氨基配体或膦配体,优选氨基配体为四甲基乙二胺、N-N’二甲基乙二胺或乙二胺,优选膦配体为三苯基膦、1,1-双(二苯基膦)二茂铁、1,2-双(二苯膦)乙烷、1,3-双(二苯膦)丙烷或三环己基膦。
进一步地,上述氰基化反应在溶剂中进行,优选溶剂为二甲基甲酰胺、N-甲基吡咯烷酮、N,N-二乙基甲酰胺、二甲基亚砜、甲苯、1,4-二氧六烷或乙腈,优选溶剂与芳基化合物的体积比为5~10:1。
进一步地,上述氰基化反应在60~100℃范围内进行。
进一步地,上述氰源中的氰根与芳基化合物的摩尔比1:1~1.6:1。
进一步地,上述催化剂与芳基化合物的摩尔比为0.005:1~0.2:1,优选为0.02:1~0.1:1。
进一步地,上述催化剂与配体的摩尔比为1:1~1:10,优选为1:1~1:3。
进一步地,上述还原剂为锌粉,锌粉与芳基化合物的摩尔比为0.05:1~1:1,优选为0.1:1~0.5:1。
应用本发明的技术方案,上述制备方法采用具有通式II的芳基化合物为底物,该物质相对于芳基卤代物的制备较为容易,因此可以降低制备芳基腈类化合物的成本;且该物质的活性比现有技术中常用的芳基底物高,因此在氰基化反应过程中,所需的反应温度较低,进一步降低了制备方法对能量的消耗,也从该方面降低了合成成本;另外,由于该物质活性较高,因此对催化剂的要求相对降低,使得一些非贵金属催化剂的使用成为可能,进而又进一步降低合成成本;进一步地,由于该物质活性较高,因此其转化率较高,那么降低了产物的分离和回收工艺的操作繁复性和复杂程度,进而还可以降低合成成本。同时,具有通式II的芳基化合物中芳基为各种芳杂环时,其反应活性与苯基时的反应活性相当,因此对于合成芳基腈类化合物具有普适性。即使得本申请的制备方法通用性强,对于含有富电子或缺电子取代基的芳基或杂芳基底物,均能获得较高的收率。
附图说明
构成本申请的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
图1示出了根据本发明实施例1的步骤一得到的目标产物的1H NMR谱图;
图2示出了根据本发明实施例1的步骤一得到的目标产物13C NMR谱图;
图3示出了根据本发明实施例2的产物的1H NMR谱图;
图4示出了根据本发明实施例8的产物的1H NMR谱图;
图5示出了根据本发明实施例8的产物的13C NMR谱图;
图6示出了根据本发明实施例9的产物的1H NMR谱图;
图7示出了根据本发明实施例9的产物的13C NMR谱图;
图8示出了根据本专利实施例21的产物的1H NMR谱图;以及
图9示出了根据本专利实施例22的产物的1H NMR谱图。
具体实施方式
需要说明的是,在不冲突的情况下,本申请中的实施例及实施例中的特征可以相互组合。下面将参考附图并结合实施例来详细说明本发明。
如背景技术所记载的,现有技术中芳基腈类化合物的制备方法存在由于各种原因导致的成本较高的问题,为了解决该问题,本申请提供了一种芳基腈类化合物的制备方法,芳基腈类化合物具有通式I:该制备方法包括:以具有通式II的芳基化合物为底物,其中,n=0~1,X1、X2、X3和X4中在化学可接受的结构中各自独立地选自N、S、O和C中的任意一种;Y为OSO2F、OTf或OTs;R1、R2、R3和R4各自独立地选自H、烷基、芳基和卤素中的任意一种,在催化剂、还原剂和配体的催化作用下使芳基化合物与氰源进行氰基化反应,得到芳基腈类化合物。
上述制备方法采用具有通式II的芳基化合物为底物,该物质相对于芳基卤代物的制备较为容易,因此可以降低制备芳基腈类化合物的成本;且该物质的活性比现有技术中常用的芳基底物高,因此在氰基化反应过程中,所需的反应温度较低,进一步降低了制备方法对能量的消耗,也从该方面降低了合成成本;另外,由于该物质活性较高,因此对催化剂的要求相对降低,使得一些非贵金属催化剂的使用成为可能,进而又进一步降低合成成本;进一步地,由于该物质活性较高,因此其转化率较高,那么降低了产物的分离和回收工艺的操作繁复性和复杂程度,进而还可以降低合成成本。
同时,具有通式II的芳基化合物中芳基为各种芳杂环时,其反应活性与苯基时的反应活性相当,因此对于合成芳基腈类化合物具有普适性。即使得本申请的制备方法通用性强,对于含有富电子或缺电子取代基的芳基或杂芳基底物,均能获得较高的收率。
在经过上述合成后,采用现有常规的提纯方法进行提纯即可,在此不再一一列举提纯方法。
在本申请一种优选的实施例中,上述氰源选自K4Fe(CN)6、Zn(CN)2、KCN和NaCN组成的组中的一种或两种。采用上述低毒性的盐类作为氰源,其使用量大大降低,同时大大降低了反应安全风险,以及后处理反应及三废处理的风险和成本。而且上述K4Fe(CN)6的成本比其他金属氰化物成本更加低廉,合成工艺绿色环保,环境无污染。当然,如果不考虑安全问题,现有技术中常用的氰源也可以用于本申请。
如前所描述的,由于底物的反应活性提高,因此对催化剂催化活性要求可以适当降低,比如选择非贵金属盐作为催化剂,优选非贵金属盐选自CuI、CuBr、CuCl、Cu(OAc)2(醋酸铜)、Cu(acac)2(乙酰丙酮铜)、Cu(OTf)2(三氟甲烷磺酸铜)、CuI2、CuCl2、CuSO4、NiX2、Ni(OAc)2(醋酸镍)、NiX2(dppf)、NiX2(dppe)、NiX2(dppp)、Ni(PCy3)X2和Ni(Py)2Cl2组成的组中的任意一种或多种,其中X表示卤素,dppf表示1,1-双(二苯基膦)二茂铁,dppe表示1,2-双(二苯膦)乙烷,dppp表示1,3-双(二苯膦)丙烷,PCy3表示三环己基膦,Py表示吡啶。上述各非贵金属盐的成本较低,因此可以进一步降低芳基腈类化合物的合成成本。当然,现有技术中常用的钯催化剂也可以使用,比如PdCl2、Pd(OAc)2(醋酸钯)、Pd(PPh3)4(四(三苯基膦)钯)、Pd(dba)2(三(二亚苄基丙酮)二钯)、Pd(dppf)Cl2(1,1-双(二苯基膦)二茂铁二氯化钯)、Pd(acac)2(乙酰丙酮钯)。
在选用上述催化剂时,本领域技术人员可以根据现有技术知识选择合适的配体,优选上述配体为氨基配体或膦配体,进一步优选氨基配体为四甲基乙二胺(TMEDA)、N-N’二甲基乙二胺(DMEDA)或乙二胺(EDA),进一步优选膦配体为三苯基膦(PPh3)、1,1-双(二苯基膦)二茂铁(dppf)、1,2-双(二苯膦)乙烷(dppe)、1,3-双(二苯膦)丙烷dppp或三环己基膦(PCy3)。从上述配体中选择与上述催化剂相适用的种类,以进一步提高催化效率和活性。
本申请上述各配体价格低廉,且能够获得较高的体系纯度和收率,其上述催化剂配合使用后的催化效率与Pd催化剂相比,在大多数底物上获得相当或更高的收率,而上述的铜或镍催化剂价格更加低廉,大大降低生产成本,易于工艺化生产。
此外,优选上述氰基化反应在溶剂中进行,进一步优选溶剂为二甲基甲酰胺(DMF)、N-甲基吡咯烷酮(NMP)、N,N-二乙基甲酰胺、二甲基亚砜(DMSO)、甲苯、1,4-二氧六烷或乙腈。上述各溶剂为本领域的常用溶剂,其使用较为安全且成本较低。为了进一步提高反应速率,优选溶剂与芳基化合物的体积比5~10:1。以使反应底物和氰源既能充分分散接触,又能保证不至于因为分散度过大导致反应效率下降。
在本申请的底物具有较高反应活性的基础上,上述氰基化反应的反应温度得到降低,优选上述氰基化反应在60~100℃范围内进行,优选为80℃。该温度范围相对于现有技术中大于100℃的反应温度明显降低,因此对底物的稳定性要求较低,且对复杂官能团化(比如化合物结构中含有比如酯基、酮羰基或者芳环上具有卤素原子的官能团)的底物具有较好的官能团兼容性,有利于获得较高的分离收率和体系纯度;同时,其对设备和能源的要求均有明显下降,且更利于本申请制备方法在工业中的推广应用。
在本申请一种优选的实施例中,上述氰源中的氰根与芳基化合物的摩尔比为1:1~1.6:1。
此外,为了在保证催化效率的前提下,尽可能减少催化剂使用量以降低成本,优选上述催化剂与芳基化合物的摩尔比为0.005:1~0.2:1,优选为0.02:1~0.1:1。
进一步地,优选催化剂与配体的摩尔比为1:1~1:10,优选为1:1~1:3,更优选为1:2。配体与催化剂摩尔比为2:1符合催化剂的四齿配位要求,在催化循环过程中,有足够量的配体的配位和离去,而配体过多会浪费配体,增加成本。
此外,优选还原剂为锌粉,锌粉与芳基化合物的摩尔比为0.05:1~1:1,优选为0.1:1~0.5:1。利用还原剂去还原催化剂,保证反应进行;利用锌粉作为还原剂能够使反应稳定进行且成本较低;锌粉的用量控制在上述范围内,优选为0.1/1。锌粉是用来还原催化剂成零价金属,在催化循环过程中是零价金属为活性催化剂中间体。
此外,为保证降低溶剂的使用成本,优选溶剂与底物的体积比为5:1~20:1,优先为10:1。
上述各实施例中的底物可以取自现有技术中的已有产品,也可以在使用时进行合成,其合成方法也可以参考现有技术,在此不再赘述。
以下将结合实施例和对比例进一步说明本申请的有益效果。
实施例1
合成路线为:
步骤一:
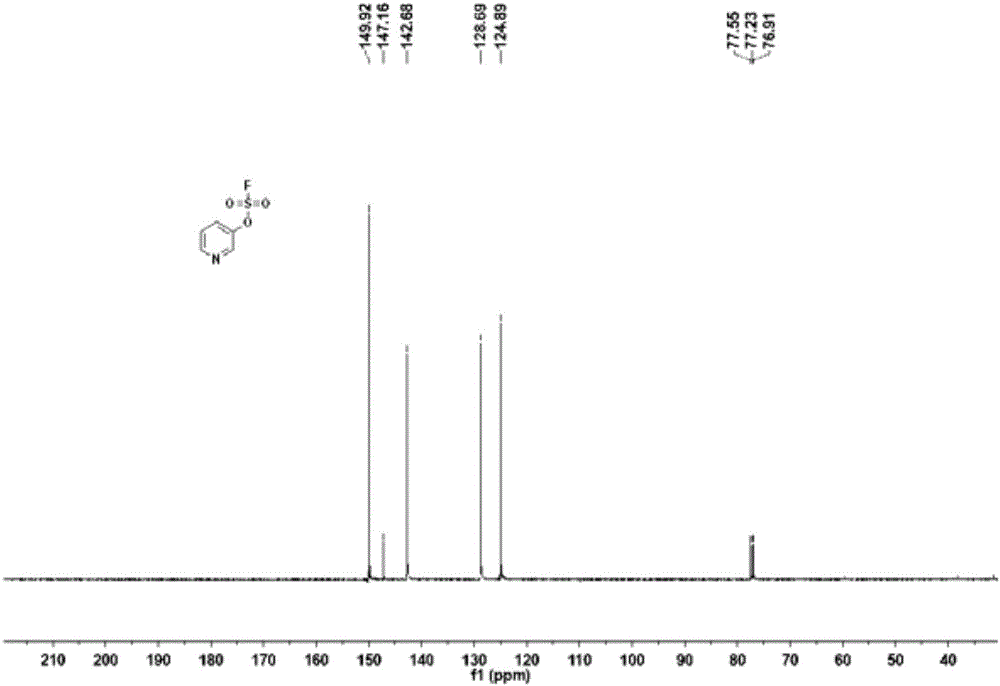
将3-羟基吡啶(100g,1.05mol)和三乙胺(160g,1.58mol)加入到二氯甲烷(500mL)中形成第一混合体系,在室温下,向该反应体系中通入磺酰氟气体(118g,1.1equiv.)并持续搅拌2~3h后,取样跟踪至原料消失后,反应体系降温到0-5℃,向反应体系加入0℃冰水300g淬灭,DCM萃取(每次300mLDCM,共三次)后,有机相合并浓缩,进行柱层析,所用层析柱中石油醚和乙酸乙酯体积比为5/1,得到油状液体176g,目标产物收率95%。
产物验证:1H NMR(400MHz,CDCl3)δ8.61(s,2H),7.69–7.60(m,1H),7.38(dd,J=8.5,4.8Hz,1H).;13C NMR(100MHz,CDCl3)δ149.92,147.16,142.68,128.69,124.89,
步骤二
将步骤一得到的目标产物作为底物,取10g(56.5mmol),催化剂Ni(dppf)Cl2(1.93g,2.8mmol),配体dppf(3.13g,5.56mmol),锌粉(0.367g,5.65mmol),氰源氰化锌(4.23g,45.2mmol)加入到50mL DMF中,得到第二混合体系,该第二混合体系氮气置换后,控制氧含量小于0.03%,将第二混合体系升温到80℃,并在80℃下搅拌3h后TLC跟踪至原料反应完毕后得到第二产物体系,将第二产物体系降温到室温后,加入MTBE(60mL)和10%氨水(130mL)并混合形成第三混合体系,第三混合体系静置后分液,所得水相再次用MTBE(45mL*2)萃取,所得有机相加入10%氨水(20mL)洗涤后分液再次得到有机相,将上述过程得到的有机相合并,然后用浓度为4mol/L的HCl调节有机相的pH到1~2后,分液,所得水相用质量分数为30%NaOH调节pH到7~8搅拌1~2小时后,测试水相pH为7~8,则直接过滤析出的固体,固体烘干得5.0g白色固体,产物收率95%。
产物验证:1H NMR(500MHz,CDCl3)δ8.68,8.68,7.85,7.83,7.82,7.69,7.67,7.53,7.52,7.51,7.51,7.50.ESI-MS Calcd for C6H5N2[M+H]+:105.1;found for 105.1。
实施例2
将实施例1的步骤一得到的目标产物作为底物,取10g(56.5mmol),催化剂Ni(dppf)Cl2(1.93g,2.8mmol),配体PPh3(1.46g,5.56mmol),锌粉(0.367g,5.65mmol),氰源氰化锌(4.23g,45.2mmol)加入到50mL DMF中,得到第二混合体系,得到第二混合体系,该第二混合体系氮气置换后,控制氧含量小于0.03%,将第二混合体系升温到80℃,并在80℃下搅拌3h后TLC跟踪至原料反应完毕后得到第二产物体系,将第二产物体系降温到室温后,加入MTBE(60mL)和10%氨水(130mL)并混合形成第三混合体系,第三混合体系静置后分液,所得水相再次用MTBE(45mL*2)萃取,所得有机相加入10%氨水(20mL)洗涤后分液再次得到有机相,将上述过程得到的有机相合并,然后用浓度为4mol/L的HCl调节有机相的pH到1~2后,分液,所得水相用质量分数为30%NaOH调节pH到7~8搅拌1~2小时后,测试水相pH为7~8,则直接过滤析出的固体,固体烘干得5.51g白色固体,产物收率94%。
产物验证:1H NMR(500MHz,CDCl3)δ8.68,8.68,7.85,7.83,7.82,7.69,7.67,7.53,7.52,7.51,7.51,7.50.ESI-MS Calcd for C6H5N2[M+H]+:105.1;found for 105.1。
实施例3
将实施例1的步骤一得到的目标产物作为底物,取10g(56.5mmol),催化剂Ni(dppf)Cl2(0.77g,1.13mmol),配体dppf(1.28g,2.26mmol),锌粉(0.367g,5.65mmol),氰源氰化锌(4.23g,45.2mmol)加入到50mL DMF中,得到第二混合体系,该第二混合体系氮气置换后,控制氧含量小于0.03%,将第二混合体系升温到80℃,并在80℃下搅拌3h后TLC跟踪至原料反应完毕后,将体系降温到15-25℃后,加入MTBE(60mL)和10%氨水(130mL)体系静置后分液,所得水相再次用MTBE(45mL*2)萃取,所得有机相加入10%氨水(20mL)洗涤后分液再次得到有机相,将上述过程得到的有机相合并,将上述过程得到的有机相合并浓缩后柱层析,洗脱剂石油醚/乙酸乙酯=10/1(体积比),得到油状液体5.57g,收率95%。
产物验证:1H NMR(500MHz,CDCl3)δ8.68,8.68,7.85,7.83,7.82,7.69,7.67,7.53,7.52,7.51,7.51,7.50.ESI-MS Calcd for C6H5N2[M+H]+:105.1;found for 105.1。
实施例4
将实施例1的步骤一得到的目标产物作为底物,取10g(56.5mmol),催化剂Ni(dppf)Cl2(0.19g,0.28mmol),配体dppf(0.31g,0.565mmol),锌粉(0.367g,5.65mmol),氰源氰化锌(4.23g,45.2mmol)加入到50mL DMF中,得到第二混合体系,该第二混合体系氮气置换后,控制氧含量小于0.03%,将第二混合体系升温到80℃,并在80℃下搅拌3h后TLC跟踪至原料反应完毕后,将体系降温到15-25℃后,加入MTBE(60mL)和10%氨水(130mL)体系静置后分液,所得水相再次用MTBE(45mL*2)萃取,所得有机相加入10%氨水(20mL)洗涤后分液再次得到有机相,将上述过程得到的有机相合并,将上述过程得到的有机相合并浓缩后柱层析,洗脱剂石油醚/乙酸乙酯=10/1(体积比),得到油状液体5.16,收率88%。
产物验证:1H NMR(500MHz,CDCl3)δ8.68,8.68,7.85,7.83,7.82,7.69,7.67,7.53,7.52,7.51,7.51,7.50.ESI-MS Calcd for C6H5N2[M+H]+:105.1;found for 105.1。
实施例5
将实施例1的步骤一得到的目标产物作为底物,取10g(56.5mmol),催化剂Ni(dppp)Cl2(0.15g,0.28mmol),配体dppp(0.23g,0.565mmol),锌粉(0.367g,5.65mmol),氰源氰化锌(4.23g,45.2mmol)加入到50mL DMF中,得到第二混合体系,该第二混合体系氮气置换后,控制氧含量小于0.03%,将第二混合体系升温到80℃,并在80℃下搅拌3h后TLC跟踪至原料反应完毕后,将体系降温到15-25℃后,加入MTBE(60mL)和10%氨水(130mL)体系静置后分液,所得水相再次用MTBE(45mL*2)萃取,所得有机相加入10%氨水(20mL)洗涤后分液再次得到有机相,将上述过程得到的有机相合并,将上述过程得到的有机相合并浓缩后柱层析,洗脱剂石油醚/乙酸乙酯=10/1(体积比),得到油状液体5.57g,收率95%。
产物验证:1H NMR(500MHz,CDCl3)δ8.68,8.68,7.85,7.83,7.82,7.69,7.67,7.53,7.52,7.51,7.51,7.50.ESI-MS Calcd for C6H5N2[M+H]+:105.1;found for 105.1。
实施例6
将实施例1的步骤一得到的目标产物作为底物,取10g(56.5mmol),催化剂Ni(dppp)Cl2(0.15g,0.28mmol),配体PPh3(0.15g,0.565mmol),锌粉(0.367g,5.65mmol),氰源氰化锌(4.23g,45.2mmol)加入到50mL DMF中,得到第二混合体系,该第二混合体系氮气置换后,控制氧含量小于0.03%,将第二混合体系升温到80℃,并在80℃下搅拌3h后TLC跟踪至原料反应完毕后,将体系降温到15-25℃后,加入MTBE(60mL)和10%氨水(130mL)体系静置后分液,所得水相再次用MTBE(45mL*2)萃取,所得有机相加入10%氨水(20mL)洗涤后分液再次得到有机相,将上述过程得到的有机相合并,将上述过程得到的有机相合并浓缩后柱层析,洗脱剂石油醚/乙酸乙酯=10/1(体积比),得到油状液体5.2g,收率88%。
产物验证:1H NMR(500MHz,CDCl3)δ8.68,8.68,7.85,7.83,7.82,7.69,7.67,7.53,7.52,7.51,7.51,7.50.ESI-MS Calcd for C6H5N2[M+H]+:105.1;found for 105.1。
实施例7
将实施例1的步骤一得到的目标产物作为底物,取10g(56.5mmol),催化剂Pd(PPh3)4(1.31g,1.13mmol),配体PPh3(0.592g,2.26mmol),锌粉(0.367g,5.65mmol),氰源氰化锌(4.23g,45.2mmol)加入到50mL DMF中,得到第二混合体系,该第二混合体系氮气置换后,控制氧含量小于0.03%,将第二混合体系升温到80℃,并在80℃下搅拌3h后TLC跟踪至原料反应完毕后,将体系降温到15-25℃后,加入MTBE(60mL)和10%氨水(130mL)体系静置后分液,所得水相再次用MTBE(45mL*2)萃取,所得有机相加入10%氨水(20mL)洗涤后分液再次得到有机相,将上述过程得到的有机相合并,将上述过程得到的有机相合并浓缩后柱层析,洗脱剂石油醚/乙酸乙酯=10/1(体积比),得到油状液体5.28g,收率90%。
产物验证:1H NMR(500MHz,CDCl3)δ8.68,8.68,7.85,7.83,7.82,7.69,7.67,7.53,7.52,7.51,7.51,7.50.ESI-MS Calcd for C6H5N2[M+H]+:105.1;found for 105.1。
实施例8
将4-苯甲酰基硫酰氟作为底物,取10g(35.7mmol),催化剂Ni(dppf)Cl2(0.488g,0.714mmol),配体dppf(0.792g,1.43mmol),锌粉(0.232g,3.57mmol),氰源氰化锌(3.35g,28.56mmol)加入到50mL DMF中,得到第二混合体系,该第二混合体系氮气置换后,控制氧含量小于0.03%,将第二混合体系升温到80℃,并在80℃下搅拌3h后TLC跟踪至原料反应完毕后,将体系降温到15-25℃后,加入MTBE(60mL)和10%氨水(130mL)体系静置后分液,所得水相再次用MTBE(45mL*2)萃取,所得有机相加入10%氨水(20mL)洗涤后分液再次得到有机相,将上述过程得到的有机相合并,将上述过程得到的有机相合并浓缩后柱层析,洗脱剂石油醚/乙酸乙酯=10/1(体积比),得到白色固体6.6g,收率89%。
1H NMR(400MHz,CDCl3)δ7.89(s,1H),7.88(s,1H),7.81(m,2H),7.79(m,2H),7.67-7.63(m,1H),7.54(m,2H).13C NMR(100MHz,CDCl3)δ195.17,141.39,136.50,133.48,132.33,130.39,130.22,128.80,118.17,115.82.ESI-MS Calcd for C14H10NO[M+H]+:208.1;found for 208.1
实施例9
将8-氟磺酰酯基喹啉作为底物,取10g(44.1mmol),催化剂Ni(dppf)Cl2(0.60g,0.88mmol),配体dppf(0.98g,1.76mmol),锌粉(0.287g,4.41mmol),氰源氰化锌(4.13g,35.3mmol)加入到50mL DMF中,得到第二混合体系,该第二混合体系氮气置换后,控制氧含量小于0.03%,将第二混合体系升温到80℃,并在80℃下搅拌3h后TLC跟踪至原料反应完毕后,体系降温到15~25℃后,加入MTBE(60mL)和10%氨水(130mL)淬灭体系,静置后分液,所得水相再次用MTBE(45mL*2)萃取,所得有机相加入10%氨水(20mL)洗涤后分液再次得到有机相,将上述过程得到的有机相合并浓缩后柱层析,洗脱剂石油醚/乙酸乙酯=3/1(体积比),得到白色固体6.2g收率91%。
1H NMR(400MHz,CDCl3)δ9.09(dd,J=4.2,1.4Hz,1H),8.25(dd,J=8.3,1.5Hz,1H),8.12(dd,J=7.2,0.7Hz,1H),8.08(d,J=8.3Hz,1H),7.62(t,J=7.7Hz,1H),7.56(dd,J=8.3,4.3Hz,1H).13C NMR(100MHz,CDCl3)δ152.67,147.65,136.66,135.69,133.08,128.30,126.04,122.95,117.41,113.29;ESI-MS Calcd for C10H7N2[M+H]+:155.1;found for 155.1。
实施例10
将实施例1的步骤一得到的目标产物作为底物,取10g(56.5mmol),催化剂Ni(dppf)Cl2(1.93g,2.8mmol),配体dppf(3.13g,5.56mmol),锌粉(0.367g,5.65mmol),氰源氰化钾(5.88g,90.4mmol)加入到50mL DMF中,得到第二混合体系,该第二混合体系氮气置换后,控制氧含量小于0.03%,将第二混合体系升温到60℃,并在60℃下搅拌3h后TLC跟踪至原料反应完毕后,将体系降温到15-25℃后,加入MTBE(60mL)和10%氨水(130mL)体系静置后分液,所得水相再次用MTBE(45mL*2)萃取,所得有机相加入10%氨水(20mL)洗涤后分液再次得到有机相,将上述过程得到的有机相合并,将上述过程得到的有机相合并浓缩后柱层析,洗脱剂石油醚/乙酸乙酯=10/1(体积比),得到油状液体5.22g,收率89%。
产物验证:1H NMR(500MHz,CDCl3)δ8.68,8.68,7.85,7.83,7.82,7.69,7.67,7.53,7.52,7.51,7.51,7.50.ESI-MS Calcd for C6H5N2[M+H]+:105.1;found for 105.1。
实施例11
将实施例1的步骤一得到的目标产物作为底物,取10g(56.5mmol),催化剂Ni(dppf)Cl2(1.93g,2.8mmol),配体dppf(3.13g,5.56mmol),锌粉(0.367g,5.65mmol),氰源氰化钠(4.43g,90.4mmol)加入到50mL DMF中,得到第二混合体系,该第二混合体系氮气置换后,控制氧含量小于0.03%,将第二混合体系升温到100℃,并在100℃下搅拌3h后TLC跟踪至原料反应完毕后,将体系降温到15-25℃后,加入MTBE(60mL)和10%氨水(130mL)体系静置后分液,所得水相再次用MTBE(45mL*2)萃取,所得有机相加入10%氨水(20mL)洗涤后分液再次得到有机相,将上述过程得到的有机相合并,将上述过程得到的有机相合并浓缩后柱层析,洗脱剂石油醚/乙酸乙酯=10/1(体积比),得到油状液体5.34g,收率91%。
产物验证:1H NMR(500MHz,CDCl3)δ8.68,8.68,7.85,7.83,7.82,7.69,7.67,7.53,7.52,7.51,7.51,7.50.ESI-MS Calcd for C6H5N2[M+H]+:105.1;found for 105.1。
实施例12
将实施例1的步骤一得到的目标产物作为底物,取10g(56.5mmol),催化剂Cu(acac)2(0.73g,2.8mmol),配体TMEDA(0.645g,5.56mmol),锌粉(0.367g,5.65mmol),氰源氰化锌(5.28g,45.2mmol)加入到50mL DMF中,得到第二混合体系,该第二混合体系氮气置换后,控制氧含量小于0.03%,将第二混合体系升温到80℃,并在80℃下搅拌3h后TLC跟踪至原料反应完毕后,将体系降温到15-25℃后,加入MTBE(60mL)和10%氨水(130mL)体系静置后分液,所得水相再次用MTBE(45mL*2)萃取,所得有机相加入10%氨水(20mL)洗涤后分液再次得到有机相,将上述过程得到的有机相合并,将上述过程得到的有机相合并浓缩后柱层析,洗脱剂石油醚/乙酸乙酯=10/1(体积比),得到油状液体5.16g,收率88%。
产物验证:1H NMR(500MHz,CDCl3)δ8.68,8.68,7.85,7.83,7.82,7.69,7.67,7.53,7.52,7.51,7.51,7.50.ESI-MS Calcd for C6H5N2[M+H]+:105.1;found for 105.1。
实施例13
将实施例1的步骤一得到的目标产物作为底物,取10g(56.5mmol),催化剂Ni(dppf)Cl2(1.93g,2.8mmol),配体dppf(3.13g,5.56mmol),锌粉(0.367g,5.65mmol),氰源氰化锌(3.97g,33.9mmol)加入到50mL DMF中,得到第二混合体系,该第二混合体系氮气置换后,控制氧含量小于0.03%,将第二混合体系升温到80℃,并在80℃下搅拌3h后TLC跟踪至原料反应完毕后,将体系降温到15-25℃后,加入MTBE(60mL)和10%氨水(130mL)体系静置后分液,所得水相再次用MTBE(45mL*2)萃取,所得有机相加入10%氨水(20mL)洗涤后分液再次得到有机相,将上述过程得到的有机相合并,将上述过程得到的有机相合并浓缩后柱层析,洗脱剂石油醚/乙酸乙酯=10/1(体积比),得到油状液体5.4g,收率92%。
产物验证:1H NMR(500MHz,CDCl3)δ8.68,8.68,7.85,7.83,7.82,7.69,7.67,7.53,7.52,7.51,7.51,7.50.ESI-MS Calcd for C6H5N2[M+H]+:105.1;found for 105.1。
实施例14
将实施例1的步骤一得到的目标产物作为底物,取10g(56.5mmol),催化剂Ni(dppf)Cl2(3.86g,5.6mmol),配体dppf(9.39g,16.7mmol),锌粉(0.367g,5.65mmol),氰源氰化锌(5.28g,45.2mmol)加入到50mL DMF中,得到第二混合体系,该第二混合体系氮气置换后,控制氧含量小于0.03%,将第二混合体系升温到80℃,并在80℃下搅拌3h后TLC跟踪至原料反应完毕后,将体系降温到15-25℃后,加入MTBE(60mL)和10%氨水(130mL)体系静置后分液,所得水相再次用MTBE(45mL*2)萃取,所得有机相加入10%氨水(20mL)洗涤后分液再次得到有机相,将上述过程得到的有机相合并,将上述过程得到的有机相合并浓缩后柱层析,洗脱剂石油醚/乙酸乙酯=10/1(体积比),得到油状液体5.2g,收率89%。
产物验证:1H NMR(500MHz,CDCl3)δ8.68,8.68,7.85,7.83,7.82,7.69,7.67,7.53,7.52,7.51,7.51,7.50.ESI-MS Calcd for C6H5N2[M+H]+:105.1;found for 105.1。
实施例15
将实施例1的步骤一得到的目标产物作为底物,取10g(56.5mmol),催化剂CuBr(0.40g,2.8mmol),配体DMEDA(0.49g,5.6mmol),锌粉(0.367g,5.65mmol),氰源氰化锌(5.28g,45.2mmol)加入到50mL DMF中,得到第二混合体系,该第二混合体系氮气置换后,控制氧含量小于0.03%,将第二混合体系升温到80℃,并在80℃下搅拌3h后TLC跟踪至原料反应完毕后,将体系降温到15-25℃后,加入MTBE(60mL)和10%氨水(130mL)体系静置后分液,所得水相再次用MTBE(45mL*2)萃取,所得有机相加入10%氨水(20mL)洗涤后分液再次得到有机相,将上述过程得到的有机相合并,将上述过程得到的有机相合并浓缩后柱层析,洗脱剂石油醚/乙酸乙酯=10/1(体积比),得到油状液体4.93g,收率84%。
产物验证:1H NMR(500MHz,CDCl3)δ8.68,8.68,7.85,7.83,7.82,7.69,7.67,7.53,7.52,7.51,7.51,7.50.ESI-MS Calcd for C6H5N2[M+H]+:105.1;found for 105.1。
实施例16
将实施例1的步骤一得到的目标产物作为底物,取10g(56.5mmol),催化剂CuSO4(0.45g,2.8mmol),配体PPy3(1.56g,5.6mmol),锌粉(0.367g,5.65mmol),氰源氰化锌(5.28g,45.2mmol)加入到50mL甲苯中,得到第二混合体系,该第二混合体系氮气置换后,控制氧含量小于0.03%,将第二混合体系升温到80℃,并在80℃下搅拌3h后TLC跟踪至原料反应完毕后,将体系降温到15-25℃后,加入MTBE(60mL)和10%氨水(130mL)体系静置后分液,所得水相再次用MTBE(45mL*2)萃取,所得有机相加入10%氨水(20mL)洗涤后分液再次得到有机相,将上述过程得到的有机相合并,将上述过程得到的有机相合并浓缩后柱层析,洗脱剂石油醚/乙酸乙酯=10/1(体积比),得到油状液体4.75g,收率81%。
产物验证:1H NMR(500MHz,CDCl3)δ8.68,8.68,7.85,7.83,7.82,7.69,7.67,7.53,7.52,7.51,7.51,7.50.ESI-MS Calcd for C6H5N2[M+H]+:105.1;found for 105.1。
实施例17
将实施例1的步骤一得到的目标产物作为底物,取10g(56.5mmol),催化剂Ni(OAc)2(0.199g,1.13mmol),配体TMEDA(0.26g,2.3mmol),锌粉(0.367g,5.65mmol),K4Fe(CN)6(17.93g,16.9mmol)加入到50mL N,N-二乙基苯甲酰胺中,得到第二混合体系,该第二混合体系氮气置换后,控制氧含量小于0.03%,将第二混合体系升温到80℃,并在80℃下搅拌3h后TLC跟踪至原料反应完毕后,将体系降温到15-25℃后,加入MTBE(60mL)和10%氨水(130mL)体系静置后分液,所得水相再次用MTBE(45mL*2)萃取,所得有机相加入10%氨水(20mL)洗涤后分液再次得到有机相,将上述过程得到的有机相合并,将上述过程得到的有机相合并浓缩后柱层析,洗脱剂石油醚/乙酸乙酯=10/1(体积比),得到油状液体5.0g,收率86%。
产物验证:1H NMR(500MHz,CDCl3)δ8.68,8.68,7.85,7.83,7.82,7.69,7.67,7.53,7.52,7.51,7.51,7.50.ESI-MS Calcd for C6H5N2[M+H]+:105.1;found for 105.1。
实施例18
将实施例1的步骤一得到的目标产物作为底物,取10g(56.5mmol),催化剂Ni(Py)2Cl2(0.325g,1.13mmol),配体DMEDA(0.206g,2.3mmol),锌粉(1.84g,28.3mmol),K4Fe(CN)6(17.93g,16.9mmol)加入到50mL NMP中,得到第二混合体系,该第二混合体系氮气置换后,控制氧含量小于0.03%,将第二混合体系升温到80℃,并在80℃下搅拌3h后TLC跟踪至原料反应完毕后,将体系降温到15-25℃后,加入MTBE(60mL)和10%氨水(130mL)体系静置后分液,所得水相再次用MTBE(45mL*2)萃取,所得有机相加入10%氨水(20mL)洗涤后分液再次得到有机相,将上述过程得到的有机相合并,将上述过程得到的有机相合并浓缩后柱层析,洗脱剂石油醚/乙酸乙酯=10/1(体积比),得到油状液体4.7g,收率80%。
产物验证:1H NMR(500MHz,CDCl3)δ8.68,8.68,7.85,7.83,7.82,7.69,7.67,7.53,7.52,7.51,7.51,7.50.ESI-MS Calcd for C6H5N2[M+H]+:105.1;found for 105.1。
实施例19
将实施例1的步骤一得到的目标产物作为底物,取10g(56.5mmol),催化剂Ni(dppf)Cl2(7.7g,11.3mmol),配体dppf(12.8g,22.6mmol),锌粉(3.67g,56.5mmol),氰源氰化锌(4.23g,45.2mmol)加入到50mL DMF中,得到第二混合体系,该第二混合体系氮气置换后,控制氧含量小于0.03%,将第二混合体系升温到80℃,并在80℃下搅拌3h后TLC跟踪至原料反应完毕后,将体系降温到15-25℃后,加入MTBE(60mL)和10%氨水(130mL)体系静置后分液,所得水相再次用MTBE(45mL*2)萃取,所得有机相加入10%氨水(20mL)洗涤后分液再次得到有机相,将上述过程得到的有机相合并,将上述过程得到的有机相合并浓缩后柱层析,洗脱剂石油醚/乙酸乙酯=10/1(体积比),得到油状液体5.45g,收率93%。
产物验证:1H NMR(500MHz,CDCl3)δ8.68,8.68,7.85,7.83,7.82,7.69,7.67,7.53,7.52,7.51,7.51,7.50.ESI-MS Calcd for C6H5N2[M+H]+:105.1;found for 105.1。
实施例20
将实施例1的步骤一得到的目标产物作为底物,取10g(56.5mmol),催化剂Ni(dppf)Cl2(0.77g,1.13mmol),配体dppf(1.28g,2.26mmol),锌粉(0.184g,2.83mmol),氰源氰化锌(4.23g,45.2mmol)加入到50mL DMF中,得到第二混合体系,该第二混合体系氮气置换后,控制氧含量小于0.03%,将第二混合体系升温到80℃,并在80℃下搅拌3h后TLC跟踪至原料反应完毕后,将体系降温到15-25℃后,加入MTBE(60mL)和10%氨水(130mL)体系静置后分液,所得水相再次用MTBE(45mL*2)萃取,所得有机相加入10%氨水(20mL)洗涤后分液再次得到有机相,将上述过程得到的有机相合并,将上述过程得到的有机相合并浓缩后柱层析,洗脱剂石油醚/乙酸乙酯=10/1(体积比),得到油状液体5.34g,收率91%。
产物验证:1H NMR(500MHz,CDCl3)δ8.68,8.68,7.85,7.83,7.82,7.69,7.67,7.53,7.52,7.51,7.51,7.50.ESI-MS Calcd for C6H5N2[M+H]+:105.1;found for 105.1。
实施例21
2-乙酰-5-磺酰氟噻吩为底物,取9.9g(44.1mmol),催化剂Ni(dppf)Cl2(0.60g,0.88mmol),配体dppf(0.98g,1.76mmol),锌粉(0.287g,4.41mmol),氰源氰化锌(4.13g,35.3mmol)加入到50mL DMF中,得到第二混合体系,该第二混合体系氮气置换后,控制氧含量小于0.03%,将第二混合体系升温到80℃,并在80℃下搅拌3h后TLC跟踪至原料反应完毕后,体系降温到15~25℃后,加入MTBE(60mL)和10%氨水(130mL)淬灭体系,静置后分液,所得水相再次用MTBE(45mL*2)萃取,所得有机相加入10%氨水(20mL)洗涤后分液再次得到有机相,将上述过程得到的有机相合并浓缩后柱层析,洗脱剂石油醚/乙酸乙酯=3/1(体积比),得到白色固体6.1g收率92%。
1H NMR(500MHz;CDCl3):δ7.63(d,J=4.0Hz,1H),7.61(d,J=4.0Hz,1H),2.60(s,3H);13C NMR(125MHz;CDCl3):δ189.9,149.9,137.8,131.3,116.4,113.4,27.2
实施例22
7-磺酰氟-4-甲基香豆素为底物,取11.38g(44.1mmol),催化剂Ni(dppf)Cl2(0.60g,0.88mmol),配体dppf(0.98g,1.76mmol),锌粉(0.287g,4.41mmol),氰源氰化锌(4.13g,35.3mmol)加入到50mL DMF中,得到第二混合体系,该第二混合体系氮气置换后,控制氧含量小于0.03%,将第二混合体系升温到80℃,并在80℃下搅拌3h后TLC跟踪至原料反应完毕后,体系降温到15~25℃后,加入MTBE(60mL)和10%氨水(130mL)淬灭体系,静置后分液,所得水相再次用MTBE(45mL*2)萃取,所得有机相加入10%氨水(20mL)洗涤后分液再次得到有机相,将上述过程得到的有机相合并浓缩后柱层析,洗脱剂石油醚/乙酸乙酯=3/1(体积比),得到白色固体7.58g收率93%。
1H NMR(500MHz;CDCl3):δ7.71(d,J=8.2Hz,1H),7.61(d,J=1.6Hz,1H),7.57(dd,J=8.1,1.6Hz,1H),6.43(q,J=1.4Hz,1H),2.47(d,J=1.3Hz,3H);13C NMR(125MHz;CDCl3):δ159.2,153.2,151.0,127.5,125.8,123.7,120.9,117.9,117.5,115.0,18.8
经过上述各实施例的合成过程及结果可以看出,在反应温度较低的情况下,仍然能够取得较高的转化率;且所采用的催化剂为非贵金属催化剂且用量较少时,也能取得理想的催化效果,同时得到较高转化率;进一步地,由于转化率较高,那么本申请采用上述常规的提纯方法,也能取得80%以上的收率。
从以上的描述中,可以看出,本发明上述的实施例实现了如下技术效果:
上述制备方法采用具有通式II的芳基化合物为底物,该物质相对于芳基卤代物的制备较为容易,因此可以降低制备芳基腈类化合物的成本;且该物质的活性比现有技术中常用的芳基底物高,因此在氰基化反应过程中,所需的反应温度较低,进一步降低了制备方法对能量的消耗,也从该方面降低了合成成本;另外,由于该物质活性较高,因此对催化剂的要求相对降低,使得一些非贵金属催化剂的使用成为可能,同时也可以减少催化剂的用量,进而又进一步降低合成成本;进一步地,由于该物质活性较高,因此其转化率较高,那么降低了产物的分离和回收工艺的操作繁复性和复杂程度,进而还可以降低合成成本。
同时,具有通式II的芳基化合物中芳基为各种芳杂环时,其反应活性与苯基时的反应活性相当,因此对于合成芳基腈类化合物具有普适性。即使得本申请的制备方法通用性强,对于含有富电子或缺电子取代基的芳基或杂芳基底物,均能获得较高的收率。
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!