一种喜树碱磷酸酯类化合物、其制备方法及应用与流程
本发明属于生物医药领域,更具体地,涉及一种喜树碱磷酸酯类化合物、其制备方法及应用。
背景技术:
喜树碱是Wall等于1966年首先从中国特有的珙桐科植物喜树(camptotheca acuminata)中分离得到的一种具有显著细胞毒活性的喹啉类生物碱(J.Nat.Prod.2004,67,129-135),对骨癌、肝癌、膀胱癌和白血病等多种恶性肿瘤表现出较好的抑制作用,但在临床使用时发现,喜树碱在发挥其抗肿瘤活性时会产生骨髓抑制、呕吐和腹泻等较严重的副作用,同时因其分子结构中喹啉环上氮的特殊碱性而水溶性较差,不能直接人体非肠道给药。为进一步改善其水溶性和降低其毒副作用,20世纪70年代初期对喜树碱的水溶性钠盐进行了Ⅰ期临床试验,虽然观察到了一定的抗癌活性和大大提高了该类药物的水溶性,却因其严重而且不可预见的毒副作用使得进一步临床试验中断。近年来,国内外研究者对喜树碱类药物进行了系统深入的研究,其中以其为先导衍生合成的多个活性化合物如伊立替康、拓扑替康、9-氨基喜树碱、9-硝基喜树碱、DX-8951f、GG-211、BNP-1350、ST-1481和CKD-602等已被FDA批准上市或处于临床研究阶段(①Bioorg.Med.Chem.2004,12,1585–1604;②Phytochem.2004,65,2735–2749)。但由于喜树碱类衍生物结构中具有抗肿瘤活性的内酯环(E环)在人体内生理条件下容易水解开环,造成抗肿瘤活性降低。且实验证明,在血浆中内酯环形式与开环形式存在平衡,内酯环形式的浓度和药物疗效存在正比关系。而开环形式是导致不良反应的原因所在,如引起骨髓抑制、呕吐、腹泻和血尿等((1)药学学报.2003,38,715–720;(2)J.Org.Chem.,2000,65,4601-4606。)。因此对E环-20位的结构修饰和改造,提高内酯环形式在人血浆中的比例以期提高该类化合物的活性并减小毒性,成为目前研发喜树碱类药物的主要研究课题之一。
技术实现要素:
针对现有技术的以上缺陷或改进需求,本发明提供了一种喜树碱磷酸酯类化合物、其制备方法及应用,其目的在于通过以喜树碱构效关系研究为指导,将喜树碱20位羟基进行修饰形成酯键,提高内酯环的稳定性,由此解决现有喜树碱衍生物作为抗肿瘤药物的活性低、毒副作用大的技术问题。
为实现上述目的,按照本发明的一个方面,提供了一种喜树碱磷酸酯类化合物,其特征在于,其具有式(I)的结构:
其中,R1和R2各自独立地为H或烃基,所述烃基为直链烷基、支链烷基、芳基或杂芳基。
优选地,所述R1和R2各自独立地为C1-C12的烷基。
优选地,所述R1和R2各自独立地为C1-C7的烷基。
优选地,所述R1为H、甲基、2-丙基、异丁基、苄基或2-甲基丙基;所述R2为甲基、乙基或苯基。
优选地,所述喜树碱磷酸酯类化合物是由喜树碱氨基酸酯与甲醛和亚磷酸酯反应得到。
按照本发明的另一个方面,提供了一种所述的喜树碱磷酸酯类化合物的制备方法,按照如下步骤进行:以喜树碱氨基酸酯为原料,以四氢呋喃为溶剂,向反应体系加入三乙胺和催化剂量的无水三氯化铁混合均匀,然后向反应体系加入甲醛与亚磷酸酯,所述喜树碱氨基酸酯与甲醛和亚磷酸酯反应摩尔比为1:5:2.5,在60℃~100℃反应完全后,即可得到所述的喜树碱磷酸酯类化合物。
优选地,所述制备方法还包括分离步骤,所述分离步骤为柱层析分离。
优选地,所述柱层析采用的硅胶为200~300目的柱层析用硅胶。
按照本发明的另一个方面,提供了一种所述的喜树碱磷酸酯类化合物的应用,应用于制备治疗癌症细胞的药物。
优选地,所述癌症细胞包括人肝癌细胞(Hep3B)、人肺腺癌细胞(A549)、人乳腺癌细胞株(MDA-MB-231和MCF-7)、人口腔表皮样癌细胞(KB)或人口腔表皮样癌细胞耐药株(KBvin)肿瘤细胞。
总体而言,通过本发明所构思的以上技术方案与现有技术相比,能够取得下列有益效果:
(1)本发明以喜树碱构效关系研究为指导,以不同氨基酸取代的喜树碱衍生物作为原料,将喜树碱20位羟基进行修饰形成酯键,制备得到了本发明所述的喜树碱磷酸酯类化合物,进一步阻止羟基和邻位酯羰基形成了分子内氢键,提高了内酯环的稳定性,从而提高了喜树碱的成药性、抗肿瘤活性并降低了其毒性。
(2)本发明利用氨基酸在体内被主动转运,可作为药动团连于药效团,以利于吸收和转运这一原理,同时运用“多样性合成”这一思想,将磷酸酯这一药物分子上常用的活性官能团与喜树碱氨基酸酯通过多组分反应的方式结合而合成了本发明所述的一类新型的喜树碱衍生物。
(3)经体外抗肿瘤活性筛选结果表明,本发明所述的喜树碱磷酸酯类化合物对人肺腺癌细胞(A549)、人乳腺癌细胞株(MDA-MB-231)、人口腔表皮样癌细胞(KB)和人口腔表皮样癌细胞耐药株(KBvin)表现出较强的抑制活性,且一些化合物高于目前临床药物拓扑替康,因此本发明的化合物可用于制备抗肿瘤的药物。
(4)本发明所述的喜树碱类化合物结构新颖、合成工艺简单、产品纯度高,对肿瘤细胞表现出较强的抑制作用,具有优良的应用前景。
附图说明
图1是本发明提供的喜树碱磷酸酯类化合物的通式结构图;
图2是化合物Ig处理Hep3B和MCF7细胞后的细胞表型结果示意图;
图3是化合物Ig使Hep3B和MCF7细胞周期阻滞在S/G2期并产生凋亡测试结果示意图;
图4是化合物Ig和CPT-11在试管和细胞内抑制拓扑异构酶I的活性测试结果示意图;
图5是化合物Ig在裸鼠移植瘤模型中抗肿瘤作用研究结果示意图;
图6是原位肝癌中,化合物Ig抑制肝癌形成测试结果示意图;
图7是化合物Ig在小鼠原位癌模型中抑制细胞增殖、阻滞细胞周期和促进细胞凋亡测试结果示意图;
图8是化合物Ig急性毒理研究的实验结果示意图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。此外,下面所描述的本发明各个实施方式中所涉及到的技术特征只要彼此之间未构成冲突就可以相互组合。
本发明所述的喜树碱磷酸酯类化合物具有式(I)所示的化学结构:
其中,R1和R2各自独立地为H或烃基,所述烃基为直链烷基、支链烷基、芳基或杂芳基,R1和R2各自独立地优选为C1-C12的烷基,进一步优选为C1-C7的烷基。
更进一步地优选,R1为H、甲基、2-丙基、异丁基、苄基或2-甲基丙基;所述R2为甲基、乙基或苯基。
本发明所述的喜树碱磷酸酯类化合物制备方法按如下化学反应式进行:
本发明的喜树碱磷酸酯类化合物(式I化合物)制备方法是以喜树碱氨基酸酯为原料,适量的四氢呋喃作溶剂,滴加三乙胺,向反应体系加入催化剂量的无水三氯化铁,待搅拌10分钟后,向反应体系加入反应量的甲醛与亚磷酸酯,60~100℃反应8个小时,薄层层析检测,柱层析(洗脱体系为氯仿:甲醇)分离,得到目标化合物。
本发明的喜树碱磷酸酯类化合物优选的制备方法是将喜树碱氨基酸酯(2mmol)于圆底烧瓶中,加入四氢呋喃(10mL)作溶剂,滴加三乙胺(2mmol),向反应体系加入无水三氯化铁(0.3mmol),待搅拌10分钟后,向反应体系加入甲醛(10mmol)和的亚磷酸酯(5mmol),60摄氏度反应8个小时,薄层层析板检测反应进度,待反应完成后,用饱和碳酸氢钠水溶液洗涤有机相,无水硫酸镁干燥后减压浓缩,柱层析分离(CHCl3:MeOH)纯化即得目标产物,其中柱层析采用的硅胶为200~300目的柱层析用硅胶。
本发明所用的原料喜树碱-20-O-L-氨基酯的制备方法参见文献方法(Bioorg.Med.Chem.,1998,6,551-562)。
按照本发明所述的喜树碱磷酸酯类化合物的制备方法,合成了如图1所示的通式的喜树碱磷酸酯类化合物。以下实施例给出了Ia-Ip一共16中喜树碱磷酸酯类化合物的制备合成方法,采用NMR测定了各化合物结构,并计算分析了其收率。
本发明所述的的喜树碱磷酸酯类化合物的应用,其特征在于,应用于制备治疗癌症细胞的药物,所述癌症细胞包括人肝癌细胞(Hep3B)、人肺腺癌细胞(A549)、人乳腺癌细胞株(MDA-MB-231和MCF-7)、人口腔表皮样癌细胞(KB)或人口腔表皮样癌细胞耐药株(KBvin)肿瘤细胞。
以下为实施例:
实施例1:目标化合物Ia的合成
原料喜树碱20-O-L-甘氨酸酯的合成:取N-Boc-甘氨酸(3.13mmol)于圆底烧瓶中,加入干燥的二氯甲烷(200mL)使其溶解,随后在冰浴下依次加入喜树碱(3.13mmol)、N,N'-二异丙基碳二亚胺(DIPC,3.13mmol)以及4-二甲氨基吡啶(DMAP,3.13mmol)。反应在室温下搅拌16小时。薄层层析检测反应完成后,抽滤除去固体杂质,随后用0.1N的盐酸洗涤有机相,干燥,减压浓缩后得到白色固体,柱层析(氯仿-甲醇)分离得到中间体喜树碱-20-O-(N’-叔丁氧羰基)-L-甘氨酸酯。取喜树碱-20-O-(N’-叔丁氧羰基)-L-甘氨酸酯(2mmol)于圆底烧瓶中,加入CH2Cl2(2mL)和TFA(2mL),室温搅拌2小时后减压除去溶剂,用甲醇重结晶得到最终的喜树碱-20-O-L-甘氨酸酯的三氟乙酸盐。合成方法参见文献方法(Bioorg.Med.Chem.,1998,6,551-562)。
其反应式为:
化合物Ia的合成:取喜树碱甘氨酸酸酯(2mmol)于圆底烧瓶中,加入四氢呋喃(10mL)作溶剂,随后滴加三乙胺(2mmol),之后向反应体系加入无水三氯化铁(0.3mmol),待搅拌10分钟后,向反应体系加入甲醛(10mmol)和的亚磷酸二甲酯(5mmol),60摄氏度反应8个小时,薄层层析板检测反应进度,待反应完成后,用饱和碳酸氢钠水溶液洗涤有机相,无水硫酸镁干燥后减压浓缩,柱层析分离(CHCl3-MeOH)纯化即得目标产物,其中层析用硅胶柱采用200~300目的柱层析用硅胶。
其反应式如下:
反应所得产物检测数据如下:产率:53%;熔点:285-288℃;1H-NMR(400MHz,CDCl3)δ:8.40(s,1H,C7-H),8.22(m,2H,C9-H,C12-H),7.82(t,1H,C10-H,J=4Hz),7.66(t,1H,C11-H,J=4Hz),7.25(s,1H,C14-H),5.70(d,1H,C17-H,J=16Hz),5.41(d,1H,C17-H,J=16Hz),5.31(s,2H,C5-H),3.93-4.10(m,2H,甘氨酸-H),3.74-3.80(m,9H,OMe-H),3.27-3.30(m,4H,NCH2P),2.14-2.28(m,2H,C18-H),0.97(t,3H,C19-H,J=8Hz);MS-ESI m/z:650.1[M+H]+,证实为化合物Ia。
实施例2:目标化合物Ib的合成
与实施例1同,仅以丙氨酸代替甘氨酸。反应所得产物检测数据如下:产率:51%;熔点:291-293℃;1H-NMR(400MHz,CDCl3)δ:8.40(s,1H,C7-H),8.23(m,2H,C9-H,C12-H),7.82(t,1H,C10-H,J=4Hz),7.67(t,1H,C11-H,J=4Hz),7.22(s,1H,C14-H),5.70(d,1H,C17-H,J=20Hz),5.41(d,1H,C17-H,J=20Hz),5.30(s,2H,C5-H),4.46-4.48(m,1H,丙氨酸-H),3.72-3.84(m,9H,OMe-H),3.30-3.35(m,4H,NCH2P),2.18-2.33(m,2H,C18-H),1.35(d,3H,丙氨酸-H,J=8Hz)1.01(t,3H,C19-H,J=8Hz);MS-ESI m/z:664.1[M+H]+,证实为化合物Ib。
实施例3:目标化合物Ic的合成
与实施例1同,仅以缬氨酸代替甘氨酸。反应所得产物检测数据如下:产率:56%;熔点:289-291℃;1H-NMR(400MHz,CDCl3)δ:8.40(s,1H,C7-H),8.23(m,2H,C9-H,C12-H),7.83(t,1H,J=4Hz,C10-H),7.66(t,1H,J=4Hz,C11-H),7.32(s,1H,C14-H),5.70(m,1H,C17-H),5.41(m,1H,C17-H),5.30(s,2H,C5-H),3.77-3.88(m,9H,OMe-H),3.68-3.73(m,4H,NCH2P),3.36-3.48(m,1H,缬氨酸-H),3.03-3.25(m,1H,缬氨酸-H),2.18-2.35(m,2H,C18-H),1.11-1.25(m,6H,缬氨酸-H)1.01(t,3H,J=8Hz,C19-H);MS-ESI m/z:692.1[M+H]+,证实为化合物Ic。
实施例4:目标化合物Id的合成
与实施例1同,仅以亮氨酸代替甘氨酸。反应所得产物检测数据如下:产率:57%;熔点:278-280℃;1H-NMR(400MHz,CDCl3)δ:8.40(s,1H,C7-H),8.23(m,2H,C9-H,C12-H),7.83(t,1H,C10-H,J=4Hz),7.67(t,1H,C11-H,J=4Hz),7.20(s,1H,C14-H),5.70(d,1H,C17-H,J=20Hz),5.41(d,1H,C17-H,J=20Hz),5.31(s,2H,C5-H),4.36(m,1H,亮氨酸-H),3.77-3.84(m,9H,OMe-H),3.69-3.72(m,4H,NCH2P),3.11-3.38(m,3H,亮氨酸-H),2.18-2.35(m,2H,C18-H),1.11-1.29(m,6H,亮氨酸-H,J=8Hz)0.88(t,3H,J=8Hz,C19-H);MS-ESI m/z:706.2[M+H]+,证实为化合物Id。
实施例5:目标化合物Ie的合成
与实施例1同,仅以异亮氨酸代替甘氨酸。反应所得产物检测数据如下:产率:52%;熔点:269-271℃;1H-NMR(400MHz,CDCl3)δ:8.40(s,1H,C7-H),8.23(m,2H,C9-H,C12-H),7.83(t,1H,C10-H,J=4Hz),7.67(t,1H,C11-H,J=4Hz),7.20(s,1H,C14-H),5.71(d,1H,C17-H,J=20Hz),5.41(d,1H,C17-H,J=20Hz),5.30(s,2H,C5-H),4.11-4.15(m,1H,异亮氨酸-H),3.76-3.85(m,9H,OMe-H),3.69-3.72(m,4H,NCH2P),3.02-3.19(m,1H,异亮氨酸-H),2.18-2.35(m,2H,C18-H),1.11-1.29(m,8H,异亮氨酸-H,J=8Hz),0.86(t,3H,J=8Hz,C19-H);MS-ESI m/z:706.2[M+H]+,证实为化合物Ie。
实施例6:目标化合物If的合成
与实施例1同,仅以苯丙氨酸代替甘氨酸。反应所得产物检测数据如下:产率:49%;熔点:289-292℃;1H-NMR(400MHz,CDCl3)δ:8.40(s,1H,C7-H),8.23(m,2H,C9-H,C12-H),7.82(t,1H,C10-H,J=4Hz),7.67(t,1H,C11-H,J=4Hz),7.21(d,2H,Ph-H,J=8Hz,),7.02(t,2H,Ph-H,J=8Hz),6.79(s,1H,C14-H),6.62(t,1H,Ph-H,J=8Hz),5.70(d,1H,C17-H,J=16Hz),5.41(d,1H,C17-H,J=16Hz),5.30(s,2H,C5-H),4.71-4.74(m,1H,苯丙氨酸-H),3.77-3.82(m,9H,OMe-H),3.64-3.67(m,2H,苯丙氨酸-H),3.31-3.49(m,4H,NCH2P),2.05-2.26(m,2H,C18-H),0.92(t,3H,J=8Hz,C19-H);MS-ESI m/z:740.2[M+H]+,证实为化合物If。
实施例7:目标化合物Ig的合成
与实施例1同,仅以亚磷酸二乙酯代替亚磷酸二甲酯。反应所得产物检测数据如下:产率:51%;熔点:291-293℃;1H-NMR(400MHz,CDCl3)δ:8.40(s,1H,C7-H),8.22(m,2H,C9-H,C12-H),7.82(t,1H,C10-H,J=4Hz),7.66(t,1H,C11-H,J=4Hz),7.28(s,1H,C14-H),5.70(d,1H,C17-H,J=16Hz),5.41(d,1H,C17-H,J=16Hz),5.31(s,2H,C5-H),4.08-4.17(m,10H,甘氨酸-H,OCH2CH3),3.25-3.27(m,4H,NCH2P),2.14-2.29(m,2H,C18-H),1.21-1.33(m,12H,OCH2CH3),0.97(t,3H,C19-H,J=8Hz);MS-ESI m/z:728.2[M+Na]+,证实为化合物Ig。
实施例8:目标化合物Ih的合成
与实施例1同,仅以亚磷酸二乙酯代替亚磷酸二甲酯,丙氨酸代替甘氨酸。反应所得产物检测数据如下:产率:57%;熔点:285-286℃;1H-NMR(400MHz,CDCl3)δ:8.40(s,1H,C7-H),8.23(m,2H,C9-H,C12-H),7.82(t,1H,C10-H,J=4Hz),7.67(t,1H,C11-H,J=4Hz),7.20(s,1H,C14-H),5.69(d,1H,C17-H,J=20Hz),5.40(d,1H,C17-H,J=20Hz),5.30(s,2H,C5-H),4.53-4.54(m,1H,丙氨酸-H),4.13-4.23(m,8H,OCH2CH3-H),3.29-3.34(m,4H,NCH2P),2.17-2.34(m,2H,C18-H),1.89(m,3H,丙氨酸-H),1.16-1.36(m,12H,OCH2CH3-H)1.01(t,3H,C19-H,J=8Hz);MS-ESI m/z:742.3[M+Na]+,证实为化合物Ih。
实施例9:目标化合物Ii的合成
与实施例1同,仅以亚磷酸二乙酯代替亚磷酸二甲酯,缬氨酸代替甘氨酸。反应所得产物检测数据如下:产率:61%;熔点:287-289℃;1H-NMR(400MHz,CDCl3)δ:8.40(s,1H,C7-H),8.23(m,2H,C9-H,C12-H),7.82(t,1H,C10-H,J=4Hz),7.67(t,1H,C11-H,J=4Hz),7.20(s,1H,C14-H),5.71(d,1H,C17-H,J=20Hz),5.41(d,1H,C17-H,J=20Hz),5.29(s,2H,C5-H),4.13-4.24(m,8H,OCH2CH3-H),3.97-3.99(m,1H,缬氨酸-H),3.37-3.49(m,4H,NCH2P),3.16-3.20(m,1H,缬氨酸-H),2.13-2.34(m,2H,C18-H),1.13-1.87(m,18H,OCH2CH3-H,缬氨酸-H),0.87(t,3H,C19-H,J=8Hz);MS-ESI m/z:760.1[M+Na]+,证实为化合物Ii。
实施例10:目标化合物Ij的合成
与实施例1同,仅以亚磷酸二乙酯代替亚磷酸二甲酯,亮氨酸代替甘氨酸。反应所得产物检测数据如下:产率:70%;熔点:267-269℃;1H-NMR(400MHz,CDCl3)δ:8.40(s,1H,C7-H),8.23(m,2H,C9-H,C12-H),7.82(t,1H,C10-H,J=4Hz),7.67(t,1H,C11-H,J=4Hz),7.20(s,1H,C14-H),5.70(d,1H,C17-H,J=20Hz),5.41(d,1H,C17-H,J=20Hz),5.31(s,2H,C5-H),4.10-4.26(m,9H,OCH2CH3-H,亮氨酸-H),3.37-3.49(m,4H,NCH2P),3.11-3.19(m,3H,亮氨酸-H),2.13-2.34(m,2H,C18-H),1.07-1.60(m,18H,OCH2CH3-H,亮氨酸-H),0.86(t,3H,C19-H,J=8Hz);MS-ESI m/z:784.2[M+Na]+,证实为化合物Ij。
实施例11:目标化合物Ik的合成
与实施例1同,仅以亚磷酸二乙酯代替亚磷酸二甲酯,异亮氨酸代替甘氨酸。反应所得产物检测数据如下:产率:48%;熔点:283-286℃;1H-NMR(400MHz,CDCl3)δ:8.40(s,1H,C7-H),8.23(m,2H,C9-H,C12-H),7.82(t,1H,C10-H,J=4Hz),7.67(t,1H,C11-H,J=4Hz),7.20(s,1H,C14-H),5.71(d,1H,C17-H,J=20Hz),5.41(d,1H,C17-H,J=20Hz),5.30(s,2H,C5-H),4.10-4.23(m,9H,OCH2CH3-H,异亮氨酸-H),3.37-3.49(m,4H,NCH2P),3.02-3.19(m,1H,异亮氨酸-H),2.13-2.34(m,2H,C18-H),1.07-1.60(m,20H,OCH2CH3-H,异亮氨酸-H),0.86(t,3H,C19-H,J=8Hz);MS-ESI m/z:784.3[M+Na]+,证实为化合物Ik。
实施例12:目标化合物Il的合成
与实施例1同,仅以亚磷酸二乙酯代替亚磷酸二甲酯,苯丙氨酸代替甘氨酸。反应所得产物检测数据如下:产率:53%;熔点:271-273℃;1H-NMR(400MHz,CDCl3)δ:8.40(s,1H,C7-H),8.23(m,2H,C9-H,C12-H),7.82(t,1H,C10-H,J=4Hz),7.67(t,1H,C11-H,J=4Hz),7.21(d,2H,Ph-H,J=8Hz,),6.99(t,2H,Ph-H,J=8Hz),6.77(s,1H,C14-H),6.62(t,1H,Ph-H,J=8Hz),5.69(d,1H,C17-H,J=20Hz),5.40(d,1H,C17-H,J=20Hz),5.26(s,2H,C5-H),4.80-4.84(m,1H,苯丙氨酸-H),4.13-4.23(m,8H,OCH2CH3-H),3.27-3.50(m,4H,NCH2P),2.92-2.95(m,2H,苯丙氨酸-H),2.04-2.25(m,2H,C18-H),1.25-1.36(m,12H,OCH2CH3-H)0.92(t,3H,C19-H,J=8Hz);MS-ESI m/z:818.3[M+Na]+,证实为化合物Il。
实施例13:目标化合物Im的合成
与实施例1同,仅以亚磷酸二苯酯代替亚磷酸二甲酯。反应所得产物检测数据如下:产率:49%;熔点:278-280℃;1H-NMR(400MHz,CDCl3)δ:8.40(s,1H,C7-H),8.22(m,2H,C9-H,C12-H),7.75(t,1H,C10-H,J=4Hz),7.64(t,1H,C11-H,J=4Hz),7.01-7.24(m,21H,C14-H,Ph-H),5.70(d,1H,C17-H,J=16Hz),5.41(d,1H,C17-H,J=16Hz),5.23(s,2H,C5-H),4.12(s,2H,甘氨酸-H),3.69-3.72(m,4H,NCH2P),2.07-2.24(m,2H,C18-H),0.91(t,3H,C19-H,J=8Hz);MS-ESI m/z:920.1[M+Na]+,证实为化合物Im。
实施例14:目标化合物In的合成
与实施例1同,仅以亚磷酸二苯酯代替亚磷酸二甲酯,丙氨酸代替甘氨酸。反应所得产物检测数据如下:产率:61%;熔点:276-279℃;1H-NMR(400MHz,CDCl3)δ:8.40(s,1H,C7-H),8.22(m,2H,C9-H,C12-H),7.75(t,1H,C10-H,J=4Hz),7.64(t,1H,C11-H,J=4Hz),7.07-7.24(m,21H,C14-H,Ph-H),5.72(d,1H,C17-H,J=16Hz),5.42(d,1H,C17-H,J=16Hz),5.28(s,2H,C5-H),4.69(s,2H,丙氨酸-H),3.64-3.90(m,4H,NCH2P),2.07-2.24(m,2H,C18-H),1.33(d,3H,丙氨酸-H,J=8Hz),0.91(t,3H,C19-H,J=8Hz);MS-ESI m/z:934.1[M+Na]+,证实为化合物In。
实施例15:目标化合物Io的合成
与实施例1同,仅以亚磷酸二苯酯代替亚磷酸二甲酯,异亮氨酸代替甘氨酸。反应所得产物检测数据如下:产率:63%;熔点:283-286℃;1H-NMR(400MHz,CDCl3)δ:8.40(s,1H,C7-H),8.22(m,2H,C9-H,C12-H),7.75(t,1H,C10-H,J=4Hz),7.64(t,1H,C11-H,J=4Hz),6.72-7.24(m,21H,C14-H,Ph-H),5.72(d,1H,C17-H,J=16Hz),5.42(d,1H,C17-H,J=16Hz),5.28(s,2H,C5-H),4.71(s,1H,异亮氨酸-H),3.64-3.90(m,7H,NCH2P,异亮氨酸-H),2.07-2.24(m,2H,C18-H),1.02-1.34(m,6H,异亮氨酸-H),0.91(t,3H,C19-H,J=8Hz);MS-ESI m/z:954.4[M+H]+,证实为化合物Io。
实施例16:目标化合物Ip的合成
与实施例1同,仅以亚磷酸二苯酯代替亚磷酸二甲酯,苯丙氨酸代替甘氨酸。反应所得产物检测数据如下:产率:57%;熔点:287-289℃;1H-NMR(400MHz,CDCl3)δ:8.40(s,1H,C7-H),8.22(m,2H,C9-H,C12-H),7.75(t,1H,C10-H,J=4Hz),7.64(t,1H,C11-H,J=4Hz),6.71-7.24(m,26H,C14-H,Ph-H),5.71(d,1H,C17-H,J=16Hz),5.42(d,1H,C17-H,J=16Hz),5.28(s,2H,C5-H),4.71-4.74(m,1H,苯丙氨酸-H),3.77-3.99(m,6H,NCH2P,苯丙氨酸-H),2.07-2.24(m,2H,C18-H),0.84(t,3H,C19-H,J=8Hz);MS-ESI m/z:988.1[M+H]+,证实为化合物Ip。
实施例17
化合物Ia-Ip的抗肿瘤活性的实验方法及结果
本发明的药理实验采用磺酰罗丹明B(Sulforhodamine B,SRB)比色法。肿瘤细胞培养选用10%胎牛血清(FBS)的RPMI-1640或DMEM培养基,将肿瘤细胞接种于96孔板,每个孔培养3-5×103个细胞,加入不同浓度的待测试目标化合物溶液。培养72小时后,每孔加入预冷的三氯乙酸溶液(50%,w/v)固定细胞,冰箱中固定30分钟。待96孔板室温下晾干后,每孔加入0.04%(w/v)的SRB染液(1%的乙酸配制,购自Sigma Chemical公司),染色30min后倒掉染液,用乙酸冲洗4次,去除未结合的染料,室温晾干。用100μL非缓冲Tris-base碱液溶解与细胞蛋白结合的染料,水平摇床上振荡20min,采用ELx800吸收光酶标仪(美国Bio-TeK公司生产,操作软件Gen5)测定515nm处光吸收值。所有试验设3个平行组或重复3次。化合物Ia-Ip的细胞毒活性试验结果见表1。
表1化合物Ia-IIIi的细胞毒活性试验结果:IC50(μM)
注:⑴筛选方法:磺酰罗丹明B比色法;⑵作用时间:72小时;(3)化合物编号Ia-Ip分别为前述实施例1至实施例16所得产物。
对六种肿瘤细胞体外细胞毒活性测试结果显示,本发明所合成的化合物均表现出良好的毒杀人肝癌细胞(Hep3B)、人肺腺癌细胞(A549)、人乳腺癌细胞株(MDA-MB-231和MCF-7)、人口腔表皮样癌细胞(KB)和人口腔表皮样癌细胞耐药株(KBvin)肿瘤细胞的活性,几乎所有的化合物表现出高于喜树碱临床药物伊立替康(CPT-11)的体外细胞毒活性,而在Hep3B和MCF-7细胞中所有化合物均表现出良好的抗肿瘤活性。
实施例18
化合物Ig杀伤细胞机制研究的实验方法及结果
本发明选取化合物Ig作为代表化合物,研究化合物Ia-IIIi杀伤肿瘤细胞的作用机制和分子机理。首先分别在白光和DAPI染色后用显微镜观察细胞表型,然后用流式细胞仪检测细胞周期并用western blot方法检测周期相关蛋白和凋亡相关蛋白的表达,最后检测拓扑异构酶I的活性。具体实验方法如下:
用四个浓度的化合物Ig(0,0.25,0.5和1μM)处理Hep3B细胞和MCF-7细胞48h后,直接用倒置相差显微镜在白光下观察细胞或者用DAPI染色后观察细胞核。DAPI染色步骤为用4%的多聚甲醛室温固定20min后,用含0.1%Triton X-100的PBS溶液室温孵育3min,然后用DAPI染色3分钟,再用PBS洗两次。每组细胞均在相同曝光时间下拍照,最后用ImageJ2X对细胞核大小进行分析。
为检测细胞周期,将Hep3B和MCF-7细胞接种于6孔板,每孔2×105个。从第二天开始,在不同的时间点加入0.5μM化合物Ig(0,12,24,36和48h)。处理48h后,倒掉培养基并用胰酶消化后收集细胞,然后用70%乙醇在4℃固定过夜。之后用PBS洗涤2次,加入200μL生理盐水重悬细胞,及5μL RNA酶(Rnase,10mg/mL)和2μL碘化丙啶(PI,5mg/mL)。室温孵育30min后,用流式细胞仪(美国BD公司生产)检测荧光。最后用流式分析软件FlowJo7.6分析细胞周期。
将Hep3B和MCF-7细胞接种于6孔板,每孔4×105个。从第二天开始,在不同的时间点加入0.5μM化合物Ig或5μM化合物CPT-11(0,12和18h)。处理24h后,倒掉培养基并用PBS洗涤2次。然后用加样枪完全吸净6孔板中的液体,加入配好的细胞裂解液(按照体积比100:2:1:1的RIPA:cooktail:PMSF:磷酸化蛋白酶抑制剂),每孔120μL,在冰上裂解20min。然后用超声破碎仪破碎细胞后,用预冷的低温离心机4℃、10000r/min离心5min。收集上清液体,用于western blot检测。首先配制SDS-PAGE胶,以100V、50mA的条件电泳1.5h。然后100V、275mA转膜1h,取出PVDF膜后用5%的脱脂牛奶在室温摇床的条件下封闭40min。将PVDF膜于4℃孵育一抗过夜,第二天用TBST在室温摇床上洗3次,每次5min。将PVDF膜放入相应的二抗中于室温摇床上孵育1h,再次用TBST洗3次,每次5min,最后在化学发光仪中用ECL显影成像。一抗包括cyclin A2,cylin B1,cyclin D1,cyclin E1,Bcl2,Bax和caspase-3,均以1:1000的倍数稀释。
为检测化合物Ig对拓扑异构酶I活性的影响,分别在试管水平和细胞水平进行检测。试管水平的检测是将拓扑异构酶1与vehicle、Ig(100nM和200nM)、CPT-11(100nM和200nM)在反应缓冲液(10mM Tris-HCl,pH 7.9,1mM EDTA,150mM NaCl,0.1%BSA,0.1mM亚精胺,和5%甘油)于37℃共孵育20min,然后加入250ng DNA在37℃继续反应30min,。然后加入5μL的反应终止液(0.125%溴酚蓝,25%蔗糖,和5%十二烷基肌氨酸钠)终止反应。最后将反应液在1%的琼脂糖凝胶中进行电泳,并用SYBRGreen染色后用凝胶成像系统拍照。细胞水平的检测是先用vehicle、Ig(0.25和0.5μM)或CPT-11(5μM)处理MCF7细胞6h,根据TopoGEN操作手册提取细胞核蛋白。稀释不同倍数(1、3/4/、2/3或1/2)后,用与试管水平检测拓扑异构酶1相同的方法检测拓扑异构酶1活性。
机制研究实验结果见图2,图3和图4:
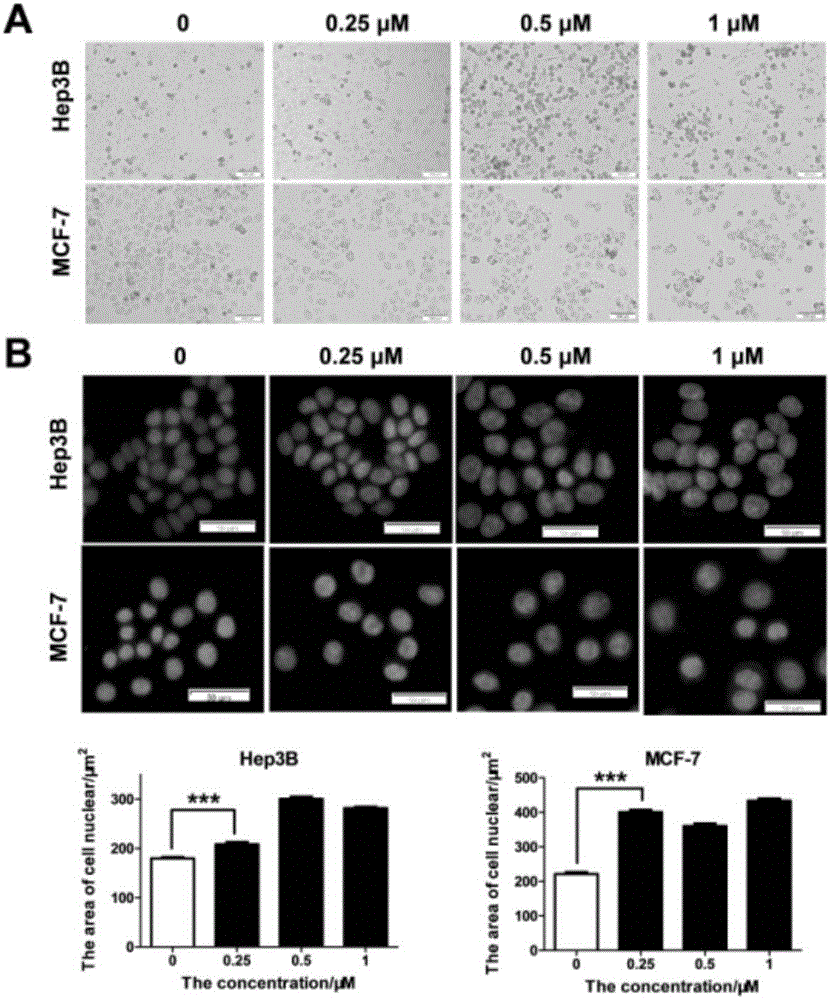
图2为化合物Ig处理Hep3B和MCF7细胞后的细胞表型。分别用不同浓度化合物Ig(0、0.25、0.5和1.0μM)处理Hep3B和MCF7细胞,48h后用倒置相差显微镜在白光下观察细胞(A);然后用DAPI染色,在倒置荧光显微镜下观察细胞核,并用imageJ2X软件统计分析细胞核大小(B)。
图3为化合物Ig使Hep3B和MCF7细胞周期阻滞在S/G2期并产生凋亡测试结果示意图。用0.5μM化合物Ig处理Hep3B和MCF7不同时间后(0、12、24、36和48h),用PI染色,然后用流式细胞仪检测细胞周期(A);用0.5μM化合物Ig或5μM CPT-11处理Hep3B和MCF7不同时间后(0、6、12和24h),用western blot检测周期和凋亡相关蛋白的表达(B)。
图4为化合物Ig和CPT-11可在试管和细胞内抑制拓扑异构酶I的活性测试结果示意图。将vehicle、化合物Ig(100和200μM)或CPT-11(100和200μM)与拓扑异构酶I孵育20min后,检测拓扑异构酶I活性(A);用vehicle、化合物Ig(0.25和0.5μM)或CPT-11(5μM)处理MCF7细胞6h后,收集核蛋白,然后稀释不同倍数(1、3/4/、2/3或1/2)检测拓扑异构酶活性(B)。R=Relaxed,S=Supercoiled。
处理48h后,随着化合物Ig浓度增加,Hep3B和MCF-7细胞明显逐渐减少,培养基中漂浮的死细胞增多(Hep3B细胞更加明显)且可见凋亡小体等凋亡现象。而用DAPI染色后,细胞核随着化合物Ig处理浓度增加而增大,在MCF-7中用1μM化合物Ig处理48h后细胞核大小约增加两倍,暗示细胞周期可能阻滞在S期或G2/M期。流式检测结果进一步表明,对照组细胞周期主要集中在G1期,用化合物Ig处理12-24h后细胞G1期减少而S期增多,36-48h后S期减少而G2/M期显著上升。说明化合物Ig确实可以使细胞周期阻滞在S/G2/M期。western blot结果与流式细胞仪检测结果一致,即在Hep3B和MCF-7细胞中随着处理时间的增加,在S/G2/M期高表达的cyclinA2和cyclinB1蛋白表达逐渐提高,而在G1期高表达的cyclinD1和cyclinE1蛋白表达逐渐降低。另外,凋亡蛋白caspase-3和Bax随着处理时间的增加而表达升高,抗凋亡蛋白Bcl2随处理时间的增加而表达下降,证明化合物Ig的确诱导Hep3B和MCF-7产生了凋亡。在试管水平上检测拓扑异构酶1活性实验表明,化合物Ig具有与CPT-11接近的拓扑异构酶1活性抑制能力,并且随着化合物处理浓度的升高,对拓扑异构酶1活性的抑制作用更加显著。在细胞水平上的拓扑异构酶1活性检测结果显示,用0.5μM化合物Ig处理过的MCF-7细胞中提取的拓扑异构酶1在稀释至3/4后即失去解螺旋能力,而用5μM化合物CPT-11处理过的MCF-7细胞中提取的拓扑异构酶1在稀释至3/4后还有很强的解螺旋能力。说明化合物Ig在试管水平具有与CPT-11接近的拓扑异构酶1抑制活性,而在细胞水平化合物Ig抑制拓扑异构酶1活性的能力比CPT-11更强。
以上实验结果表明以化合物Ig为代表的化合物Ia-Ip能够通过抑制拓扑异构酶1的活性,发挥抑制细胞增殖、阻滞细胞周期在S/G2/M期以及诱导细胞凋亡的作用。
实施例19
化合物Ig在裸鼠乳腺癌移植瘤模型中抗肿瘤作用研究的实验方法及结果
本发明选取化合物Ig作为代表化合物,研究化合物Ia-Ip在裸鼠移植瘤模型中抑制乳腺癌肿瘤生长的能力。研究方法是选取45只六周龄(约20g)雌性Balb/c裸鼠,在腋下方皮下注射2×106个MCF-7细胞。当肿瘤长至约100mm3时,尾静脉注射溶剂对照(含5%DMSO和5%氢化蓖麻油的生理盐水)、化合物Ig或CPT-11(2mg/kg),隔天注射一次,共注射7次,并且每隔3天测量体重和肿瘤大小,结果如图4所示,以2×106个MCF7注射到裸鼠皮下,等肿瘤长至约100mm3的时候开始尾静脉注射化合物Ig或CPT-11(2mg/kg),隔天注射一次,共注射7次,并每隔3天测量体重(A)和肿瘤大小(B),实验结果如图5。
由图5可以看出,裸鼠移植瘤模型中,化合物Ig可以有效抑制MCF7形成的肿瘤生长。与溶剂对照组相比,化合物Ig和CPT-11处理的小鼠体重明显较低。在25天内,溶剂对照组小鼠肿瘤体积急剧上升,而用化合物Ig和CPT-11处理小鼠的肿瘤体积增长显著减缓。说明化合物Ig与CPT-11同样具有抑制MCF-7细胞移植瘤生长的能力。该实验表明以化合物Ig为代表的化合物Ia-Ip在裸鼠乳腺癌移植瘤模型中能够起到抗肿瘤的作用。
实施例20
化合物Ig在小鼠原位癌模型中抑制肝癌形成能力研究的实验方法及结果
本发明选取化合物Ig作为代表化合物,研究化合物Ia-Ip在小鼠原位癌模型中抑制肝癌形成和发展的能力。为建立原位肝癌模型,参照文献方法将pT3/EF1α-Akt、pT-Caggs-Nrasv12和pCMV-SB三个质粒同时通过高压尾静脉注射到FVB/N小鼠体内,八周后可形成原位肝细胞肝癌。八周后,以同龄正常FVB/N作为对照组尾静脉注射溶剂对照(含5%DMSO和5%氢化蓖麻油的生理盐水),原位肝癌模型组注射溶剂对照或化合物Ig(10mg/kg),隔天注射,持续2周,期间每3天称体重。两周后,处死小鼠并解剖,观察是否有肝癌形成,然后测量肝脏大小并称重,用4%的多聚甲醛固定肝脏后做免疫组化和免疫荧光检测。免疫组化和免疫荧光的阳性率使用 Plus软件进行统计分析。免疫组化的实验步骤如下:肝脏固定过夜后先脱水,然后用二甲苯透明并浸蜡,进行石蜡包埋。将包埋的蜡块进行切片、烤片、脱蜡、水化后,将切片置于柠檬酸盐缓冲液中微波修复。冷却到室温后用PBS清洗3次,将切片放入3%的H2O2溶液中室温孵育25min以阻断内源性过氧化物酶。PBS清洗后加破膜液室温孵育10min,用1:200的山羊血清封闭,室温放置30min。将封闭好的切片每张加入100μL稀释后的一抗,覆盖组织,4℃过夜。PBS清洗后每张切片加入100μL二抗稀释液室温孵育45min。PBS清洗后每张切片加入50μL新鲜配置的DAB工作液,显微镜下控制显色。显色完全后,ddH2O冲洗。最后苏木素淡染,脱水、透明,中性树脂封片。而TUNEL免疫荧光染色是在切片与不含DNA酶的蛋白酶K在室温下反应30min后,用PBS洗3次,然后加入Tdt酶、荧光标记液和TUNEL配成的检测液于37℃避光孵育60min,再用PBS洗3次,最后封片并用荧光显微镜拍照,结果如图6和图7。
图6中将Akt/Ras/SB质粒高压尾静脉注射到FVB/N小鼠体内形成原位肝癌模型,用生理盐水高压尾静脉注射到FVB/N小鼠体内作为正常对照。八周后,正常对照组尾静脉注射溶剂对照(含5%DMSO和5%氢化蓖麻油的生理盐水),原位肝癌模型组注射溶剂对照或化合物Ig(10mg/kg),隔天注射,持续2周,期间称体重(C)。两周后,处死小鼠并解剖,观察是否有肝癌形成,然后测量肝脏大小并称重(A和B)。由图5可以看出,原位肝癌中,化合物Ig可以有效抑制肝癌形成。
图7中,将Akt/Ras/SB质粒高压尾静脉注射到FVB/N小鼠体内形成原位肝癌模型,用生理盐水高压尾静脉注射到FVB/N小鼠体内作为正常对照。八周后,正常对照组尾静脉注射溶剂对照(含5%DMSO和5%氢化蓖麻油的生理盐水),原位肝癌模型组注射溶剂对照或化合物Ig(10mg/kg),隔天注射,持续2周。两周后,处死小鼠,取肝脏用4%多聚甲醛固定,然后做免疫组化(A)和免疫荧光(B)。由图6可以看出,原位肝癌中,化合物Ig可以抑制细胞增殖、阻滞细胞周期和促进细胞凋亡。
通过检测体重,发现正常对照小鼠体重在15天内有轻微上升,原位肝癌模型组小鼠体重在11天后有明显上升,而用化合物Ig处理组小鼠体重基本维持稳定并且在第15天的体重与模型组相比有统计学差异。15天后,解剖小鼠,模型组小鼠肝脏与正常小鼠肝脏相比明显增大,并且表面变得凹凸不平,布满肿瘤结节。而用化合物Ig处理的小鼠肝脏恢复正常大小,并且表面光滑色泽鲜亮,仅有数个肿瘤结节散布(箭头所示)。肝重比结果也表明模型组肝重比增大而化合物Ig处理的小鼠肝重比恢复到正常对照组水平。H&E染色显示,正常小鼠肝脏组织质地均一、细胞排列有序、肝索明显,而模型组小鼠肝脏出现大量脂肪空泡并且组织出现不规则病变,用化合物Ig处理后小鼠的肝脏脂肪空泡明显减少并组织恢复规则。正常对照组小鼠肝脏低表达Akt,而在模型组和化合物Ig处理小鼠的肝脏中均高表达Akt,说明Akt质粒(间接也能反映Ras和SB质粒)已进入模型组和化合物Ig组小鼠肝脏并成功表达。模型组中Ki67的表达量远高于对照组说明模型组出现恶性增殖即确实已产生肿瘤,但用化合物Ig处理后Ki67的表达明显下降,表示化合物Ig能够有效抑制肿瘤细胞的恶性增殖。正常对照组中基本不表达cyclinB1是因为正常肝脏细胞不增殖,周期处于静止状态,所以周期蛋白基本不表达(包括cyclinB1)。而在模型组小鼠肝脏中cyclinB1的表达有轻微上升,是因为肝脏细胞出现恶性增殖、推动细胞周期,从而使周期蛋白表达上调。在化合物Ig处理的小鼠肝脏中cyclinB1的阳性率接近100%,该结果与化合物Ig处理肿瘤细胞的western blot结果一致,是因为化合物Ig使细胞周期大量阻滞在S/G2/M期。TUNEL免疫荧光染色显示模型组小鼠肝脏的凋亡率明显低于正常对照组小鼠,说明肝脏细胞发生肿瘤恶变后使细胞能够逃逸凋亡。而用化合物Ig处理的小鼠肝脏中凋亡率显著增加,可得到与细胞水平实验一样的结论,即化合物Ig可诱导肝癌细胞发生凋亡。
以上实验结果表明利用Akt/Ras/SB三质粒高压注射可成功建立原位肝癌模型,而化合物Ig可以通过抑制肿瘤细胞的增殖、阻滞周期S/G2/M期及促进肿瘤细胞的凋亡成功抑制该模型产生肿瘤的发生和发展。
实施例21
化合物Ig在体内急性毒性研究的实验方法及结果
为评价化合物Ig在小鼠体内的毒副作用,我们进行了急性毒理实验。选取8~10周龄的FVB/N小鼠雌性20只和雄性25只,根据体重平均分成5组,分别腹腔注射溶剂对照(含5%DMSO和5%氢化蓖麻油的生理盐水)、化合物Ig(10、50和100mg/kg)和CPT-11(100mg/kg)。此后15天内,观察老鼠死亡情况并每隔3天记录小鼠体重。15天后,眼眶取血并用抗凝管收集,然后解剖收集肝脏和肾脏并用4%的多聚甲醛固定。收集的血液一部分直接用于检测血常规,另一部分离心后收集上清,用来检测血液生化指标(肝功能指标:ALT和AST,肾功能指标:BUN和Cr)。肝脏和肾脏固定后用来做H&E染色,在显微镜下观察是否有炎性浸润等组织病变。
急性毒性实验结果见表2、表3和图8。
表2化合物Ig在雄性FVB/N小鼠急性毒性试验的血常规检测结果
表3化合物Ig在雌性FVB/N小鼠急性毒性试验的血常规检测结果
图8中将vehicle(含5%DMSO和5%氢化蓖麻油的生理盐水)、化合物Ig(10、50或100mg/kg)或CPT-11(100mg/kg)通过腹腔注射到FVB/N小鼠(雌性=4,雄性=5)体内,每3天称体重(A和C)。注射15天后,收集血液并处死小鼠,然后收集肝脏和肾脏后用4%多聚甲醛固定。血液用来检测血常规和血生化指标(B、D、E和F),肝脏和肾脏固定后用来做H&E染色(G)。由图7可以看出化合物Ig具有较低肝肾毒性。
五组小鼠在腹腔注射15天内,体重均维持在相对恒定的水平。注射15天后,血常规结果显示各组间的白细胞等与炎症相关的指标均在正常范围,其他血常规指标也未有显著异常。肝功能指标ALT和AST,及肾功能指标BUN和Cr在处理组和对照组之间也无显著差异。观察组织病理切片,肝脏和肾脏均未出现明显的炎性浸润、组织坏死及其他病变。以上结果表明,化合物Ig和CPT-11在同等高剂量下均对小鼠无明显的急性毒性。
本领域的技术人员容易理解,以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!