一种高GC含量基因突变体的创制方法与流程
本发明涉及分子生物学领域,尤其涉及一种高GC含量基因突变体的创制方法。
背景技术:
基因突变的点突变是导致生物发生遗传变异的重要物质基础之一。基因是由三联体密码子组成的,碱基的改变会引起被编码的氨基酸的改变,进而导致蛋白质结构和功能发生改变,最终影响生物表型。
生物进化过程中,通过增加基因特殊部位重要氨基酸密码子的GC含量来增强基因的稳定性,进而稳定其编码蛋白质或酶的活性功能,保证生物代谢有条不紊的进行,维持其正常的生长发育。
在分子生物学研究中,为了研究氨基酸在蛋白质或酶结构中的功能,通常采用PCR技术进行体外点突变,将野生型基因的一种氨基酸突变为另一种氨基酸,如将酸性氨基酸突变为碱性氨基酸、或极性氨基酸突变为中性氨基酸、或长链氨基酸突变为短链氨基酸、或带有基团的氨基酸突变为无基团的氨基酸等等,反之亦然。首先,将编码野生型氨基酸的密码子改变为被突变氨基酸的密码子,并以该氨基酸的密码子为中心,向上、向下各5个氨基酸密码子长度,设计相互配对的正向和反向PCR引物;根据引物Tm值,设定退火温度,在DNA聚合酶催化下,利用反应体系中的正向和反向PCR引物,合成包括载体和突变位点在内的DNA双链,最终完成基因突变体的构建。
由于设计的基因点突变引物中,在1个密码子中被突变1个碱基、或2个碱基、或3个碱基,获得相应基因突变体的难度也由此逐渐加大,尤其是获得3个碱基同时被改变的突变体的难度非常大,再加上GC含量过高,获得相应突变体就难上加难。对于一个密码子中2个碱基以上突变体的创制,在通过一次PCR反应不易获得的情况下,通常会采用间接方法,即先获得一个碱基的突变体,然后再以此突变体为模板,获得第二个碱基的突变体,以此类推,最终可能获得相应的突变体,但存在几个缺点:一是多个中间突变体的构建导致获得最终突变体所用的时间过长,而且还有可能导致基因的其它位点产生点突变,影响基因本身的功能;二是不一定能够找到构建中间突变体突变位点的合适碱基,甚至根本找不到相应的突变碱基。由于无法构建中间突变体,就只能在密码子中2个或3个碱基同时被改变的情况下直接进行PCR,但获得相应基因突变体的难度非常大。众所周知,被改变的碱基在退火过程中是无法与野生型DNA链进行正确配对的,而却与其互补引物优先配对,这就进一步增加了获取突变体的难度。由于配对碱基GC间能量高,其Tm值为3℃,而AT间能量相对较弱,其Tm值为2℃。在分子生物学操作中,经常会遇到GC含量较高的基因,即使提高退火温度,也难以获得相应基因的突变体。此外,DNA聚合酶在PCR反应中的最适温度为72℃,若因GC含量高而使退火温度超过72℃,则会影响DNA聚合酶的正常活性,不利于DNA链的延伸,也就无法获得相应的基因突变体,这就阻碍了对氨基酸及其酶蛋白功能的研究,也影响了对基因功能本身的研究。
技术实现要素:
为解决现有技术上的不足,本发明提供了一种高GC含量基因突变体的创制方法。
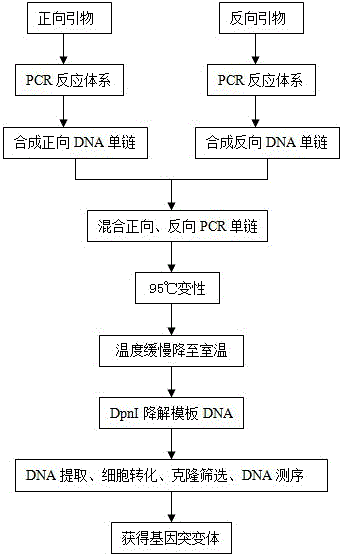
本发明基于PCR合成中,95℃下DNA变性解链,在退火温度下引物与DNA模板配对结合,在DNA聚合酶催化下合成突变位点在内DNA双链的原理,利用单引物合成DNA单链,以解决退火过程中引物自身优先配对而不能全部与DNA模板配对的问题。通过混合正向引物合成的DNA单链和反向引物合成的DNA单链,并在95℃使其充分变性解链,在温度自然缓慢下降至室温的过程中,使正向和反向单链DNA进行互补配对,DpnI酶去除模板DNA,最终创制出高GC含量的基因突变体。该技术目前在国内外尚未见报道。
本发明的技术方案如下:
本发明一种高GC含量基因突变体的创制方法,包括以下步骤:
1)将正向引物和反向引物分别加入PCR反应体系,分别合成正向DNA单链和反向DNA单链;
2)充分均匀混合含有正向DNA单链和反向DNA单链的 PCR产物;
3)95℃变性;
4)温度缓慢降至室温;
5)DpnI限制性内切酶降解模板DNA;
6)DNA提取、细菌细胞转化、克隆筛选、DNA测序,获得基因突变体。
优选的,步骤1)中所述的PCR反应体系为: 0.1μg 模板DNA,10倍pfu DNA聚合酶缓冲液5μL,10 mM dNTP 1μL,100 μM正向引物1μL,pfu DNA聚合酶(2.5U/μL)1μL、10% DMSO,加无菌水至50μL。。
所述的pfu DNA聚合酶缓冲液含有硫酸铵。
优选的步骤1)所述合成正向和反向DNA单链,PCR仪扩增程序为95℃变性0.5 分钟,66℃退火1分钟,72℃延伸13 分钟,共需要13个循环。
优选的步骤3)中所述95℃变性时间为10分钟。
优选的步骤5)中所述DpnI限制性内切酶降解模板DNA为:PCR产物混合物中加入10U/μL 的DpnI限制性内切酶2μL,混匀,37℃反应2小时或过夜,使模板DNA完全降解。
本方法适用于动物、植物、微生物以及人类等GC含量超过70%的高GC含量基因突变体的创制,或适用于改变三联体密码子中2个碱基、或3个碱基的基因突变体的创制。
本方法适用于小鼠甲羟戊酸激酶基因中丙氨酸突变为半胱氨酸的基因突变体的构建。
本发明的有益效果是:
本发明技术操作简单,所用仪器设备与常规PCR相同,可适用于动物、植物、微生物以及人类等GC含量超过70%的高GC含量基因突变体的创制,也非常适合于改变三联体密码子中2个碱基、或3个碱基的基因突变体的创制,应用前景非常广泛。
附图说明
图1 被突变的氨基酸在小鼠甲羟戊酸激酶酶结构中的位置。
图2 高GC含量基因突变体的创制流程。
具体实施方式
下面结合实施例对本发明做进一步详细的描述:
实施例1 小鼠甲羟戊酸激酶基因中丙氨酸(A)突变为半胱氨酸(C)的基因突变体的构建
突变位点:将小鼠甲羟戊酸激酶第141位的丙氨酸突变为半胱氨酸,使该酶通过自身折叠反应增加一个二硫键,从而增加酶的结构稳定性(图1)。
突变位点密码子:野生型丙氨酸的密码子为GCG;突变体半胱氨酸的密码子为TGC。突变体氨基酸密码子的3个碱基与野生型的完全不同,在退火状态下不能与野生型密码子配对。
A141C正向引物序列(A141C FPS)为:5′GAA CTG CCC CCT GGG TGC GGC TTG GGC TCC AGT G 3′;设计引物的3′端碱基必须保证为G或C。
A141C反向引物序列为(A141C RPS):5′C ACT GGA GCC CAA GCC GCA CCC AGG GGG CAG TTC3′。
引物的GC含量:整个引物包括34个碱基,涵盖11个氨基酸密码子,GC含量为73.5%,Tm值为116℃。
具体操作步骤(图2):
1.在一个灭菌的0.5 ml Eppendorf 管中,依次加入0.1μg 模板DNA,10倍pfu DNA聚合酶缓冲液(含有(NH4)2SO4 ) 5μL,10 mM dNTP 1μL,100 μM正向引物1μL,pfu DNA聚合酶(2.5U/μL)1μL、10% DMSO(二甲基亚砜),加无菌水至50μL。按照上述同样步骤在另外一个Eppendorf 管中加入上述反应物质,所不同的是正向引物换为反向引物。
2.设置PCR仪扩增程序为95℃变性0.5 分钟,66℃退火1分钟,72℃延伸13 分钟(通常每扩增1kb DNA,需要2 分钟时间。应根据目的基因及其载体的大小,设计DNA链延伸时间),共需要13个循环。
3.PCR结束后,将2个单引物扩增的PCR产物置于1个0.5ml的Eppendorf管中,充分混匀,于95℃变性10分钟,然后慢慢降温至室温。再向PCR产物混合物中加入DpnI限制性内切酶(10U/μL)2μL,混匀,37℃反应2小时或过夜,使模板DNA完全降解。
4.按常规方法进行DNA提取,并将提取、纯化的DNA溶于20 μL TE缓冲液或无菌水中。细菌细胞转化、克隆子挑选、质粒DNA提取以及DNA测序等均按常规方法进行。
以上所述仅是本专利的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本专利技术原理的前提下,还可以做出若干改进和替换,这些改进和替换也应视为本专利的保护范围。
SEQUENCE LISTING
<110> 山东省农业科学院作物研究所
<120> 一种高GC含量基因突变体的创制方法
<130> 2016
<160> 2
<170> PatentIn version 3.5
<210> 1
<211> 34
<212> DNA
<213> 人工序列
<400> 1
gaactgcccc ctgggtgcgg cttgggctcc agtg 34
<210> 2
<211> 34
<212> DNA
<213> 人工序列
<400> 2
cactggagcc caagccgcac ccagggggca gttc 34
- 还没有人留言评论。精彩留言会获得点赞!