一种快速提取革兰氏阳性菌基因组DNA的方法和试剂盒与流程
本发明涉及生物
技术领域:
,具体地,涉及一种快速提取革兰氏阳性菌基因组DNA的方法和一种快速提取革兰氏阳性菌基因组DNA的试剂盒。
背景技术:
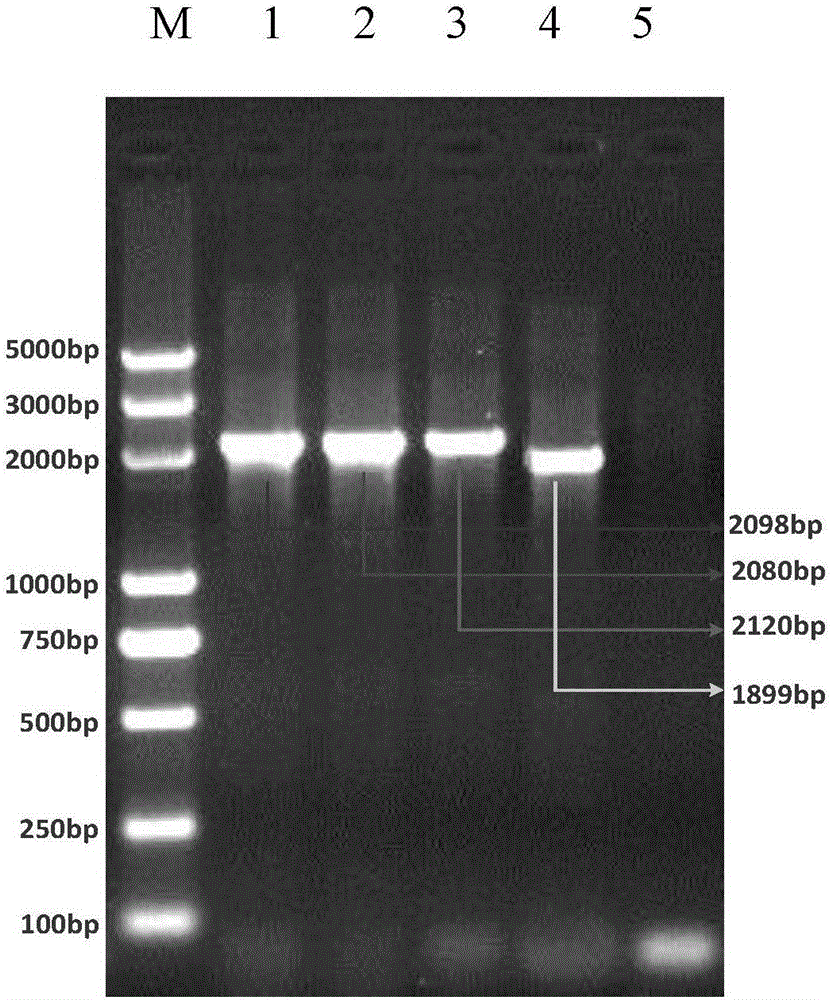
:由于革兰氏阳性菌的细胞壁相对较厚,与革兰氏阴性菌的细胞壁成分不同,给食源性致病菌检测领域带来比较大的困难,严重影响了检测效率。传统提取革兰氏阳性菌基因组的方法中,酶解法是最常用的方法。酶解法需要用酶来消化革兰氏阳性菌的细胞壁肽聚糖成分,然后用溶剂抽提,如酚/氯仿。吸附DNA材料主要采用硅质材料、阴离子交换树脂及磁珠等。该方法试剂耗材价格昂贵,成本较高。且步骤多、费时,操作过程中易产生蛋白酶残留,导致PCR反应受到抑制;酚、氯仿等有机溶剂易造成环境污染,有损实验操作者的健康。当仅有少量材料时,传统的DNA制备方法格外不适用。其他方法,如超声波法、研磨法、冻融法都能够帮助破除革兰氏阳性菌的细胞壁,但是由于操作复杂度、特殊仪器等方面的限制原因,没能够在食源性致病菌检测领域扩大使用,长期以来,革兰氏阳性菌样本中DNA的提取和纯化一直是耗时、繁琐的过程,严重减慢了检测速度,低通量的提取方法已经无法满足大量样本制备基因组DNA的需求。技术实现要素:本发明的目的是克服目前的DNA提取方法在用于革兰氏阳性细菌基因组DNA提取时所存在的效率较低的缺陷,提供一种以较高提取效率分离纯化出高收率和高纯度的革兰氏阳性细菌基因组DNA的试剂盒和方法。为了实现上述目的,本发明首先提供一种提取革兰氏阳性菌基因组DNA的试剂盒,其中,所述试剂盒包括:粒径为78μm-106μm的第一玻璃珠和粒径为425-600μm的第二玻璃珠,且所述第一玻璃珠与所述第二玻璃珠在使用时的重量比为1:(0.6-1.3)。优选的,所述第一玻璃珠与所述第二玻璃珠在使用时的重量比为1:(0.8-1.2)。本发明还提供了一种革兰氏阳性菌基因组DNA提取方法,其中,该方法包括以下步骤:(1)将菌体与DNA纯化缓冲液、粒径为78μm-106μm的第一玻璃珠和粒径为425-600μm的第二玻璃珠混合得到裂解混合物;其中,所述第一玻璃珠与所述第二玻璃珠的重量比为1:(0.6-1.3);(2)对所述裂解混合物进行脉冲震荡裂解,脉冲震荡裂解的条件为在5000-7000rpm下进行3-5次脉冲震荡,每次脉冲震荡2-4s,每次间歇1-3s获得裂解产物;(3)将所述裂解产物在80-100℃下孵育2-10min后冷却至15-25℃,离心后得到上清液。本发明还提供了所述试剂盒或提取方法在革兰氏阳性菌检测中的用途。通过上述技术方案,本发明能够实现革兰氏阳性菌基因组快速提取,克服了传统基因组提取方法上的不足之处,具备如下有益效果:(1)简便快速易操作:提取方法操作步骤少,操作简单,整个过程仅需要10分钟就可以完成。(2)提取通量高,样品需求少:可以一次性提取100个样本,检测样本可以是培养皿上的单菌落菌体或增菌液。(3)适用菌体范围广泛:采用不同粒径的玻璃珠进行组合,可以适用不同直径大小的食源性革兰氏阳性菌,提取效果可靠、高效。(4)成本低、安全可靠:不需要特殊仪器、昂贵试剂、无有毒有害物质,安全性高。本发明的其他特征和优点将在随后的具体实施方式部分予以详细说明。附图说明附图是用来提供对本发明的进一步理解,并且构成说明书的一部分,与下面的具体实施方式一起用于解释本发明,但并不构成对本发明的限制。在附图中:图1是四种革兰氏阳性菌基因组DNA琼脂糖凝胶电泳结果。其中,泳道1:金黄色葡萄球菌;泳道2:蜡样芽孢杆菌;泳道3:单增李斯特菌;泳道4:链球菌;泳道5:空白对照;M:5KMarker;图2是金黄色葡萄球菌基因组DNA的荧光定量结果;其中,A为商品化试剂盒(天根生物,DP302)提取的基因组DNA的荧光定量结果;B为用本发明试剂盒提取的基因组DNA的荧光定量结果;具体实施方式以下结合附图对本发明的具体实施方式进行详细说明。应当理解的是,此处所描述的具体实施方式仅用于说明和解释本发明,并不用于限制本发明。本发明提供了一种提取革兰氏阳性菌基因组DNA的试剂盒,其中,所述试剂盒包括:粒径为78μm-106μm的第一玻璃珠和粒径为425-600μm的第二玻璃珠,且所述第一玻璃珠与所述第二玻璃珠在使用时的重量比为1:(0.6-1.3)。其中,本发明采用的粒径为78μm-106μm的第一玻璃珠和粒径为425-600μm的第二玻璃珠混合,二者直径大小差异大,可以破碎较小(0.2μm)和较大(3μm)的细菌菌体,将二者混合使用效果明显优于单一粒径玻璃珠的破壁效果,特别适用于革兰氏阳性菌的破壁;不仅避免了由于破壁不完全造成的漏提漏检,而且显著节省了实验操作时间。其中,根据本发明一种优选的实施方式,所述第一玻璃珠与所述第二玻璃珠在使用时的重量比为1:(0.8-1.2)。在该优选实施方式中,本发明能够进一步提高样本中基因组DNA的收率和纯度。其中,所述试剂盒还含有DNA纯化缓冲液,所述DNA纯化缓冲液含有11-13%(m/v)的Chelex-100和5-15mMTris,且pH值为8.8-9.2。玻璃珠振荡破壁后细胞内容物释放到液体环境中,由于细胞内容物含有大量蛋白、糖类、以及盐离子等,这些因素会影响提取的基因组DNA的质量并进而影响下游应用的效果,例如,能够抑制PCR反应。为了能够快速的纯化DNA以满足下游应用特别是PCR反应的质量要求,本发明的技术方案采用Chelex-100和Tris的碱性溶液来纯化DNA。本发明所提供的试剂盒中,DNA纯化缓冲液在一定条件下可以导致细胞膜破裂和DNA变性,使DNA释放出来,通过高选择性地结合多价阳离子,可阻止金属离子对DNA降解的催化作用,并能去除肽类等可与DNA结合的大分子物质。进而可以快速有效的去除大多数的阳性离子,沉降大部分的蛋白,有效保护DNA。以免影响下一步的应用。所述DNA纯化缓冲液可以通过以下方法配置:用电子天平称取Chelex-100,将Chelex-100颗粒加入到无菌水中;加入1MTris(pH值为9),定容至100ml;本发明还提供了一种革兰氏阳性菌基因组DNA提取方法,其中,该方法包括以下步骤:(1)将菌体与DNA纯化缓冲液、粒径为78μm-106μm的第一玻璃珠和粒径为425-600μm的第二玻璃珠混合得到裂解混合物;其中,所述第一玻璃珠与所述第二玻璃珠的重量比为1:(0.6-1.3);(2)对所述裂解混合物进行脉冲震荡裂解,脉冲震荡裂解的条件为在5000-7000rpm下进行3-5次脉冲震荡,每次脉冲震荡2-4s,每次间歇1-3s获得裂解产物;(3)将所述裂解产物在80-100℃下孵育2-10min后冷却至15-25℃,离心后得到上清液。其中,所述DNA纯化缓冲液含有11-13%(m/v)的Chelex-100和5-15mMTris,且pH值为8.8-9.2。在本发明的一种实施方式中,所述菌体从菌液中分离得到,可以先将含有菌体的菌液离心分离得到菌体,再按照本发明所提供的方法进行基因组DNA提取。其中,在步骤(1)中可以将菌体与DNA纯化缓冲液充分混合后再加入所述第一玻璃珠和第二玻璃珠。其中,根据本发明一种优选的实施方式,所述第一玻璃珠与所述第二玻璃珠在使用时的重量比为1:(0.8-1.2)。在该优选实施方式中,本发明能够进一步提高样本中基因组DNA的收率和纯度。其中,所述脉冲震荡可以在高频旋涡振荡器上进行。玻璃珠频繁碰撞挤压产生细胞剪切作用。在本发明所提供的震荡条件下可以避免基因组DNA碎片的产生,获得高完整度的基因组DNA。在本发明的一种优选的实施方式中,在步骤(1)中,DNA裂解缓冲液的用量为0.2-1ml;第一玻璃珠和第二玻璃珠的总用量为0.15-0.25g。优选的,在步骤(3)中,离心的条件为在8000-14000g下离心1-10min。本发明还提供了所述试剂盒或提取方法在革兰氏阳性菌检测中的用途,所述用途包括提取革兰氏阳性菌的基因组DNA后进行PCR检测、基因测序检测或分子杂交检测等。以下,通过实施例进一步详细说明本发明。下述实施例中所使用的材料、试剂等,如无特殊说明,均可从商业途径得到。例如,粒径为78μm-106μm的第一玻璃珠购自Sigma公司,货号为G8893;粒径为425-600μm的第二玻璃珠购自Sigma公司,货号为G8772;Chelex-100购自Sigma公司,货号为C7901。实施例11)按照以下配方制作用于DNA纯化的材料:取4种革兰氏阳性菌:金黄色葡萄球菌、蜡样芽孢杆菌、单核细胞增生李斯特氏菌、链球菌,分别挑取单菌落,分别接种于5mLLB液体培养基的试管中培养,37℃,200rpm摇菌8h至菌体浊度为1OD。2)按照以下配方制作用于破碎细胞壁的材料:①DNA纯化缓冲液:用电子天平称取12gChelex-100,将Chelex-100颗粒加入到80ml无菌水中;②加入1MTris(pH值为9)1ml,定容至100ml;③玻璃珠A:≤106μm玻璃珠:0.1g;④玻璃珠B:425μm-600μm玻璃珠:0.1g。3)操作步骤:(1)取1.0ml菌液于EP管中,10000g离心2min;(2)去掉上清,用300μL的DNA纯化缓冲液悬浮菌体,加入0.1g玻璃珠A和0.1g玻璃珠B;(3)在涡旋振荡器上,6000rpm振荡3s,间歇2s,振荡3-5次;(4)沸水浴5min;(5)取出并放至25℃后,10000g离心1min;(6)小心吸取上清液50μL转移到新的EP管中保存,该上清含有基因组DNA。Nanodrop检测基因组DNA的浓度如下表1所示。表1菌名浓度260/280金黄色葡萄球菌82ng/μL1.94蜡样芽孢杆菌98ng/μL1.81单增李斯特菌110ng/μL1.79链球菌88ng/μL2.24)PCR反应体系的配制取200μL的PCR管配置25μL的反应体系,其配置如下:2×PCRBuffer12.5μL;25xDNA聚合酶1μL;10×引物混合液2.5μL,模板2μL,去离子水7μL。5)PCR反应将PCR管放入Bio-RadC1000型PCR仪中,开启热盖后,按照如下程序进行PCR反应:95℃5min;95℃30s,60℃30s,72℃90s,30个循环;72℃5min。6)琼脂糖凝胶电泳分析PCR产物取5μL的PCR产物与1μL的6X上样缓冲液混匀,加入到琼脂糖凝胶的加样孔中。采用2%的琼脂糖凝胶,5V/cm电泳40min,使用凝胶成像仪显示扩增目标条带的大小。琼脂糖凝胶电泳结果如图1所示,由图1可知:本发明的试剂盒可以提取不同的革兰氏阳性菌,提取得到的基因组DNA纯度高,回收得到的基因组DNA量大,无需进一步纯化完全可以满足下游的分子检测实验。对比例1不同震荡模式对提取DNA效果影响对比试验按照实施例1的方法进行金黄色葡萄球菌基因组DNA的提取,区别仅在于,震荡模式分别为:(1):持续振荡模式6000rpm,持续振荡1min;(2):脉冲振荡模式6000rpm,振荡3s,间隔2s,重复20次;Nanodrop检测基因组DNA的浓度,用琼脂糖凝胶电泳分析PCR产物,用荧光定量法评估基因组DNA的提取效果,结果显示相对于实施例1的方法,持续震荡模式与多次脉冲震荡模式下提取的DNA完整性不高,不利于下游的分子检测。对比例2不同纯化试剂纯化DNA效果对比实验按照实施例1的方法进行金黄色葡萄球菌基因组DNA的提取,区别仅在于,DNA纯化缓冲液为:(1):无菌水;(2):有12%(m/v)的Chelex-100和10mMTris,且pH值为8.5;(3):有12%(m/v)的Chelex-100和10mMTris,且pH值为9.5;Nanodrop检测基因组DNA的浓度,用琼脂糖凝胶电泳分析PCR产物,用荧光定量法评估基因组DNA的提取效果,结果显示:上述方法提取的DNA中纯度较低,含有蛋白质等抑制物,不利于下游的分子检测。对比例3玻璃珠比例对提取DNA效果影响对比试验按照实施例1的方法进行金黄色葡萄球菌基因组DNA的提取,玻璃珠共添加0.2g,区别仅在于,用量比例分别为:(1)玻璃珠A:B1:9;(2)玻璃珠A:B2:8;(3)玻璃珠A:B3:7;(4)玻璃珠A:B5:6;(5)玻璃珠A:B5:5;(6)玻璃珠A:B5:3;(7)玻璃珠A:B7:3;(8)玻璃珠A:B8:2;(9)玻璃珠A:B9:1;Nanodrop检测基因组DNA的浓度,用琼脂糖凝胶电泳分析PCR产物,用荧光定量法评估基因组DNA的提取效果,结果显示:玻璃珠A与B比例为5:6、5:5和5:3时DNA的提取效果较好,当为5:5时,效果最优,其他提取的DNA完整性不高,不利于下游的分子检测。实施例2本发明的试剂盒与市场上的商品化试剂盒(天根生物,DP302)提取效果对比试验金黄色葡萄球菌接种于5mLLB液体培养基的试管中培养,调整菌液浊度至108CFU/ml,梯度稀释至103CFU/ml。分别按照实施例1的方法和对照试剂盒(天根生物,DP302)说明书给出的方法进行金黄色葡萄球菌基因组DNA的提取。Nanodrop检测108CFU/ml菌液提取的基因组DNA的浓度,用荧光定量法评估基因组DNA的提取效果。表2紫外分光光度法测的浓度试剂盒108CFU/ml浓度260/280本发明试剂盒89ng/μL1.94对照试剂盒21ng/μL1.81表3荧光定量PCR法测定提取基因组各稀释度的CT值如表2可知,本发明所提取得到的基因组浓度是对照试剂盒提取得到的4.2倍。如表3可知,根据CT大小判断,本发明提取的金黄色葡萄球菌基因组DNA浓度要明显高于对照试剂盒提取的基因组DNA浓度,与紫外分光光度法测的浓度结果相一致。且仅需10min即可完成提取,是对照试剂盒所需时间的1/6。如图2可知,本发明所提取的基因组DNA纯度和浓度完全满足荧光定量PCR的要求。本发明可以提取非常少量的样品,提取的DNA质量完全满足荧光定量PCR实验所需,本发明所能提取少量样品的效果是市面上绝大多数基因组提取试剂盒所达不到的。以上结合附图详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合,。为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其同样应当视为本发明所公开的内容。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1