一种具有可控缓释功能的分子印迹聚合物的制作方法
本发明涉及本发明涉及药物缓释技术领域,具体而言,涉及一种具有可控缓释功能的分子印迹聚合物。
背景技术:
分子印迹聚合物(molecularly imprinted polymers,MIP)是一种人工理性设计制备的特定分子识别材料,它的高效识别性对复杂样品基质中的目标分子有很好的选择富集作用。分子印迹聚合物在纯化、催化、抗体模拟等领域获得了广泛应用,但在缓释载药方面的研究和应用还是一个较新领域。
MIP用于药物缓释系统主要有两个优点:一是由于药物模板分子与MIP之间存在着多个相互作用的位点和相匹配的空穴,因此MIP对药物模板分子的吸附能力较传统的药物包裹胶囊要强,具有更长久的释药时间;二是由于MIP是为药物模板分子量身定做的,因此对释放的药物模板分子具有高度的选择性,特别是对一些手性药物分子的释放,是传统药物包裹胶囊所无法比拟的。之前有研究表明,与非分子印迹材料相比,分子印迹材料对茶碱显示出了更好的缓释特性;另一项研究以柳氮磺吡啶(一种用于治疗结肠疾病的前体药)为模板分子,用沉淀聚合法制备了柳氮磺吡啶分子印迹小球。结果表明,该印迹小球口服后在胃(pH=1.0)中不释放药物,在小肠(pH=6.0-6.8)、大肠(pH=8.3-8.4)和结肠(pH=6.5-7.5)内缓慢释药,从而达到结肠靶向给药的目的;以四环素为模板分子制备的MIP在水溶液中也显示出对四环素的缓释效应,在起始阶段,四环素从非印迹聚合物中释放的速度几乎是其从印迹聚合物中释放速度的两倍。还有研究采用沉淀聚合法合成了左旋甲基多巴分子印迹聚合物微球(MIPMs),并对分子印迹效果和药物缓释效果进行了表征,表明分子印迹微球确实具有缓释药物的效果。
然而,目前文献报道的分子印迹材料对药物的释放速率与非印迹材料差别不大,结果难以令人满意,且多以丙烯酸、丙烯酰胺等为功能单体,制得的分子印迹材料生物相容和降解性较差。如何运用MIP设计安全、有效、稳定的给药系统,增加MIP的生物相容性是分子印迹缓释载药系统需要解决的关键性问题。
有鉴于此,特提出本发明。
技术实现要素:
本发明的目的在于提供一种具有可控缓释功能的分子印迹聚合物,以解决上述问题。
为了实现本发明的上述目的,特采用以下技术方案:
一种分子印迹聚合物,所述分子印迹聚合物为以β-环糊精修饰的甲基丙烯酸作为功能单体,以硫酸阿托品为模板分子。
β-环糊精(β-Cyclodextrin,β-CD)是一类由D-吡喃葡萄糖单元构成的环状低聚糖总称,其中β-CD分子是大小适中呈圆台状,空腔外侧亲水而内部疏水,是一种新型包合材料。环糊精广泛应用于分子识别、色谱分离、化学传感器、药物释放等领域,是继冠醚之后的第二代超分子。环糊精可以作为一种具有多种优点的新型功能单体,与模板分子在一定聚合体系下合成具有特殊的空腔结构和良好的分子识别能力的聚合物。但由于β-CD本身溶解度低、对释放的药物缺乏选择性等缺点,使其应用受到制约。药物分子与β-CD修饰的甲基丙烯酸形成环糊精包合物后,药物理化学性质得到显著改善,为药物的新型制剂、药物缓释材料的发展提供了有效手段。
本发明提供的具有可控缓释功能的分子印迹聚合物,结合了分子印迹聚合物(molecularly imprinted polymers,MIP)的类特异性特征与β-环糊精的缓释功能,从而具有更加长效的释药时间;且由于MIP是为药物模板分子量身定做的,因此对释放的药物模板分子具有高度的选择性,特别是对一些手性药物分子的释放,是传统药物包裹胶囊所无法比拟的。
可选的,如上所述的分子印迹聚合物,所述β-环糊精修饰的甲基丙烯酸的制备方法包括:
1)、将甲基丙烯酸与甲苯二异氰酸酯溶于非质子极性溶剂中,在二月桂酸二丁基锡的催化下发生聚合反应;
2)、步骤1)中所得产物中补加所述非质子极性溶剂,并加入β-环糊精进行反应,将反应产物经洗涤、重结晶及干燥后既得。
可选的,如上所述的分子印迹聚合物,所述甲基丙烯酸、甲苯二异氰酸酯及β-环糊精的摩尔比为(0.4~0.6):(0.8~1.2):(0.4~0.6)。
可选的,如上所述的分子印迹聚合物,所述硫酸阿托品与所述β-环糊精修饰的甲基丙烯酸的摩尔比为1:(3~5)。
可选的,如上所述的分子印迹聚合物,所述分子印迹聚合物以三羟甲基丙烷三甲基丙烯酸酯为交联剂、偶氮二异丁腈为引发剂在非质子极性溶剂中经聚合反应后脱除模板分子而得到。
可选的,如上所述的分子印迹聚合物,所述非质子极性溶剂为二甲基乙酰胺。
可选的,如上所述的分子印迹聚合物,所述聚合反应的类型为沉淀聚合法,具体的,该方法包括:
将所述模板分子、功能单体、交联剂和引发剂溶解在非质子极性溶剂中,随后通惰性气体除氧,热引发聚合反应,再经洗脱以及干燥后即得。
可选的,如上所述的分子印迹聚合物,将所述模板分子、功能单体、交联剂和引发剂溶解在惰性溶剂中时,先将所述模板分子以及功能单体溶解在非质子极性溶剂中并预聚合25~35min后,再加入所述交联剂和引发剂。
可选的,如上所述的分子印迹聚合物,所述热引发聚合反应的反应条件为55℃~65℃聚合反应21h~25h。
可选的,如上所述的分子印迹聚合物,洗脱时所用的洗脱液为体积比为8:2的甲醇和乙酸的混合溶液。
与现有技术相比,本发明的有益效果为:
本发明提供的新型MIP对于硫酸阿托品的持续释放在医学给药方面,是一种很有潜力的候选材料,可以依靠硫酸阿托品在人体内不同给药环境的释放率来控制药物的释放时间,从而实现药物缓释作用。
附图说明
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1为本发明实验例1中阿托品负载于不同功能单体的印迹聚合物和非印迹聚合物;
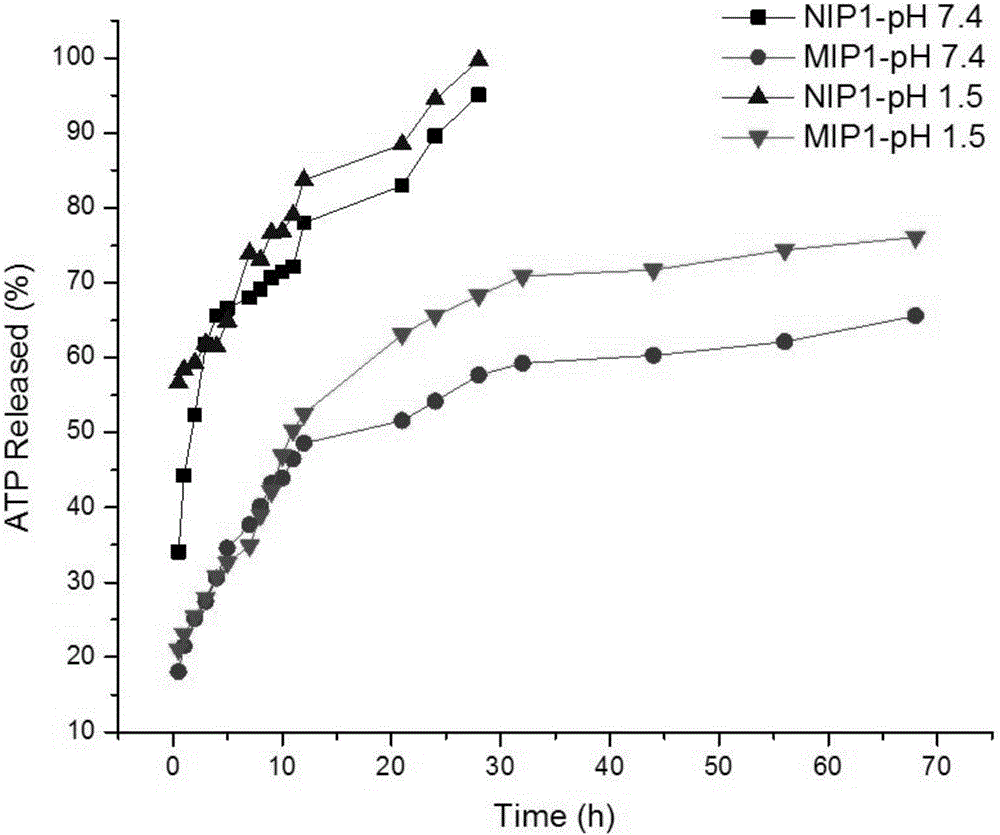
图2为本发明实验例2中ATP在模拟胃液(pH 1.5)和肠液(pH 7.4)中的释放性能。
具体实施方式
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限制本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
本发明提供的分子印迹聚合物以β-环糊精修饰的甲基丙烯酸作为功能单体,以硫酸阿托品为模板分子。
硫酸阿托品分子中具有不对称的碳原子,具有手性特征。由于MIP是为药物模板分子量身定做的,因此对释放的药物模板分子具有高度的选择性,特别是对一些手性药物分子的释放,是传统药物包裹胶囊所无法比拟的。
优选的,如上所述的分子印迹聚合物,所述β-环糊精修饰的甲基丙烯酸的制备方法包括:
1)、将甲基丙烯酸与甲苯二异氰酸酯溶于非质子极性溶剂中,在二月桂酸二丁基锡的催化下发生聚合反应;
2)、步骤1)中所得产物中补加所述非质子极性溶剂,并加入β-环糊精进行反应,将反应产物经洗涤、重结晶及干燥后既得。
优选的,如上所述的分子印迹聚合物,所述甲基丙烯酸、甲苯二异氰酸酯及β-环糊精的摩尔比为(0.4~0.6):(0.8~1.2):(0.4~0.6)。
更优选的,所述甲苯二异氰酸酯(TDI)为甲苯-2,4-二异氰酸酯。
甲基丙烯酸(Methacrylic acid,MAA)与TDI发生聚合反应后形成了分子印迹聚合物功能单体的骨架。
优选的,如上所述的分子印迹聚合物,所述硫酸阿托品与所述β-环糊精修饰的甲基丙烯酸的摩尔比为1:(3~5);
更优选的,所述硫酸阿托品与所述β-环糊精修饰的甲基丙烯酸的摩尔比为1:4。
优选的,如上所述的分子印迹聚合物,所述分子印迹聚合物以手性药物分子为模板分子。
优选的,如上所述的分子印迹聚合物,所述分子印迹聚合物以三羟甲基丙烷三甲基丙烯酸酯为交联剂、偶氮二异丁腈为引发剂在非质子极性溶剂中经聚合反应后脱除模板分子而得到。
为了使生成的印迹聚合物具有一定的空间网络结构和稳定的结合位点,分子印迹聚合物要求的交联度很高。本发明所优选的三羟甲基丙烷三甲基丙烯酸酯(TRIM)具有较高的交联度(70~90%),且在预聚合溶液中具有良好的溶解性。
分子印迹聚合反应多为自由基引发反应,因此在反应时需加入引发剂引发聚合反应,本发明优选偶氮二异丁腈(AIBN)作为引发剂。偶氮二异丁腈是油溶性的偶氮引发剂,偶氮类引发剂反应稳定,是一级反应,没有副反应,比较好控制。
优选的,如上所述的分子印迹聚合物,所述非质子极性溶剂为二甲基乙酰胺。
在沉淀聚合法制备苯丙氨酸分子印迹聚合物微球过程中,溶剂具有重要作用,主要起溶剂和致孔剂的作用。作为溶剂使模板分子、功能单体、交联剂及引发剂溶解;作为致孔剂,使得到的分子印迹聚合物有合适的孔径,使得模板分子自由与结合位点结合。通常选择的溶剂要具备以下3个条件:1)、能溶解模板分子和功能单体;2)、能形成大的流通孔;3)、对模板分子与功能单体之间的相互作用干扰小。本发明所采用的二甲基乙酰胺(Dimethylacetamide)是一种强极性非质子化溶剂,能溶解多种化合物,与水、醚、酮、酯等完全互溶,具有热稳定性高、不易水解、腐蚀性低、毒性小等特点。
本领域技术人员可根据本发明提供的分子印迹聚合物、模板分子、功能单体、交联剂种类选用本体聚合法、沉淀聚合法、悬浮聚合法、电聚合法;最优选的,采用沉淀聚合法,具体的,该方法包括:
将所述模板分子、功能单体、交联剂和引发剂溶解在非质子极性溶剂中,随后通惰性气体除氧,热引发聚合反应,再经洗脱以及干燥后即得。
本发明所采用的沉淀聚合法,特点是得到的产物粒径比较均一,比表面积大,具有更多的识别位点,因而具有更优秀的吸附性能。
优选的,如上所述的分子印迹聚合物,将所述模板分子、功能单体、交联剂和引发剂溶解在惰性溶剂中时,先将所述模板分子以及功能单体溶解在非质子极性溶剂中并预聚合25~35min后,再加入所述交联剂和引发剂。
优选的,如上所述的分子印迹聚合物,所述热引发聚合反应的反应条件为55℃~65℃聚合反应21h~25h;更优选的,反应条件为60℃聚合反应23h。
优选的,如上所述的分子印迹聚合物,洗脱时所用的洗脱液为体积比为8:2的甲醇和乙酸的混合溶液。
实施例1
(1)合成官能化的功能单体:
按照化学计量比为0.4M MAA:1.2M TDI:0.4Mβ-CD来合成。用甲苯-2,4-二异氰酸酯(TDI,2.284mL,0.4M),甲基丙烯酸(MAA,1.018mL,1.2M)混合溶于20mL二甲基乙酰胺(DMAC),搅拌,加0.1%(0.02mL)的二月桂酸二丁基锡(DBTDL)做催化剂。氮吹10min,室温下磁力搅拌1h。通过紫外分析MAA-TDI体系,确定反应。然后加入β-环糊精(β-CD,0.4M,454mg),补充5mL二甲基乙酰胺。氮吹,搅拌2h(TLC跟踪反应,展开剂:石油醚/乙酸乙酯,2/1~3/1,v/v)。得产物MAA-β-CD,用超纯水洗涤3次,再用甲醇重结晶得到白色固体,真空干燥至恒重。
(2)热引发沉淀聚合制备阿托品分子印迹聚合物。
将硫酸阿托品(69.5mg,0.1mmol)和MAA-β-CD(406mg,0.3mmol)分散溶解于30mL DMAC/H2O(20/10,v/v),超声波振动25min。在此溶液中加入TRIM(638μL,2mmol)和AIBN(30mg),溶解;N2脱气10min,水浴震荡反应。温度从室温缓慢升至55℃,聚合25h。甲醇洗涤聚合物微粒,离心(13000rpm,4℃,10min)分离收集。最后用甲醇/乙酸(80/20,v/v)洗脱液在索氏提取器中洗涤模板分子至HPLC检测不到ATP。MIP中剩余的乙酸分别用甲醇和超纯水洗涤。
实施例2
(1)合成官能化的功能单体:
按照化学计量比为0.6M MAA:0.8M TDI:0.6Mβ-CD来合成。用甲苯-2,4-二异氰酸酯(TDI,3.426mL,0.6M),甲基丙烯酸(MAA,0.678mL,0.8M)混合溶于20mL二甲基乙酰胺(DMAC),搅拌,加0.1%(0.02mL)的二月桂酸二丁基锡(DBTDL)做催化剂。氮吹10min,室温下磁力搅拌1h。通过紫外分析MAA-TDI体系,确定反应。然后加入β-环糊精(β-CD,0.6M,680mg),补充5mL二甲基乙酰胺。氮吹,搅拌2h(TLC跟踪反应,展开剂:石油醚/乙酸乙酯,2/1~3/1,v/v)。得产物MAA-β-CD,用超纯水洗涤4次,再用甲醇重结晶得到白色固体,真空干燥至恒重。
(2)热引发沉淀聚合制备阿托品分子印迹聚合物。
将硫酸阿托品(69.5mg,0.1mmol)和MAA-β-CD(676mg,0.5mmol)分散溶解于30mL DMAC/H2O(20/10,v/v),超声波振动35min。在此溶液中加入TRIM(638μL,2mmol)和AIBN(30mg),溶解;N2脱气10min,水浴震荡反应。温度从室温缓慢升至65℃,聚合21h。甲醇洗涤聚合物微粒,离心(13000rpm,4℃,10min)分离收集。最后用甲醇/乙酸(80/20,v/v)洗脱液在索氏提取器中洗涤模板分子至HPLC检测不到ATP。MIP中剩余的乙酸分别用甲醇和超纯水洗涤。
实施例3
(1)合成官能化的功能单体:
按照化学计量比为0.5M MAA:1M TDI:0.5Mβ-CD来合成。用甲苯-2,4-二异氰酸酯(TDI,2.855mL,0.5M),甲基丙烯酸(MAA,0.848mL,1M)混合溶于20mL二甲基乙酰胺(DMAC),搅拌,加0.1%(0.02mL)的二月桂酸二丁基锡(DBTDL)做催化剂。氮吹10min,室温下磁力搅拌1h。通过紫外分析MAA-TDI体系,确定反应。然后加入β-环糊精(β-CD,0.5M,567mg),补充5mL二甲基乙酰胺。氮吹,搅拌2h(TLC跟踪反应,展开剂:石油醚/乙酸乙酯,2/1~3/1,v/v)。得产物MAA-β-CD,用超纯水洗涤3次,再用甲醇重结晶得到白色固体,真空干燥至恒重。
(2)热引发沉淀聚合制备阿托品分子印迹聚合物。
将硫酸阿托品(69.5mg,0.1mmol)和MAA-β-CD(541mg,0.4mmol)分散溶解于30mL DMAC/H2O(20/10,v/v),超声波振动30min。在此溶液中加入TRIM(638μL,2mmol)和AIBN(30mg),溶解;N2脱气10min,水浴震荡反应。温度从室温缓慢升至60℃,聚合23h。甲醇洗涤聚合物微粒,离心(13000rpm,4℃,10min)分离收集。最后用甲醇/乙酸(80/20,v/v)洗脱液在索氏提取器中洗涤模板分子至HPLC检测不到ATP。MIP中剩余的乙酸分别用甲醇和超纯水洗涤。
实验例1分子印迹聚合物载药性能试验
对比例设置方法:对比例非印迹聚合物(NIP)的制备按照实施例3中的方法进行,不同之处仅在于,在制备过程中不加入模板分子ATP。
取不同浓度的1ml ATP(100、200、400、100、200、1000ng/ml)溶液,使其溶解2.5mg的聚合物(根据实施例3中记载的方法制备的MIP),在室温使用振荡器摇动2h。平衡后,高速离心。收集上层清液过0.22μm聚醚砜滤膜并通过LC-MS/MS检测。NIP以同样的方法操作。单位质量的水溶性聚合物吸附ATP的质量即聚合物吸附性能,与药物初始浓度有关呈线性关系。试验评价了MAA-β-CD较MAA作为单体制备MIP/NIP合成的水溶性聚合物微球在水溶液环境下对目标药物ATP的吸附性能,图1是水溶液中ATP在各聚合物吸附剂上的吸附等温线。从图可以看出,等温吸附线满足一级动力学方程,MIP1(MAA-β-CD为单体)吸附能力大于MIP0(MAA为单体);NIP1(MAA-β-CD为单体)吸附能力也大于NIP0(MAA为单体);同样,MIP1/MIP0的吸附能力优于NIP1/NIP0。这是由于MAA的羧基和β-CD中大量的羟基与ATP的羟基有更高的亲和作用,此外β-CD的桶状空腔结构也加强了聚合物在水溶液环境中对包含物的吸附性和选择特异性。结果表明,MIP微球对阿托品的负载率远远高于NIP负载率,因此,本发明制备的分子印迹聚合物具有很好的载药量。
实验例2阿托品分子印迹聚合物的可控释放性能
将50mg充分负载ATP的MIP微球的分别浸入到10L模拟胃液(pH=1.5)和肠液(pH=7.4)中;体系在在37℃的水浴震荡器中以180rpm/min充分震荡。在0.5到68h内的时间间隔里,离心取1mL上清液,并补充1mL新的模拟胃液或肠液继续实验。通过LC-MS/MS进行分析检测。以同样的方式以NIP作对照实验。
如图2所示,在模拟胃液和肠液的条件下,NIP在模拟胃肠液中有一个显著的突释现象,并且NIP在前3h内累积释放量可达到61.9%,这与同等条件下的MIP相比高出33.9%,NIP1中的载药量在最初的12h内释放速度非常快,并且在28h内几乎可以达到完全释放;此外,NIP对ATP释放性能与pH变化并不敏感。对于MIPs,ATP在模拟胃液中的释放(11h达到50%)比在肠液中的释放(21h接近或少于50%)更快,MIP在一定的时间间隔内的ATP释放率要低于NIP,这表明ATP和MIP的结合位点之间有特异性相互作用。因此,本发明制备的MIP具有很好的酸碱度可控释放性能。基于以上结论,新型的MIP对于ATP的持续释放在医学给药方面,是一种很有潜力的候选材料,可以依靠ATP在人体内不同给药环境的释放率来控制药物的释放时间,从而实现药物缓释作用。
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;为叙述方便,本发明仅在实验例中给出了最佳实施例3的效果,但实际上在本申请权利要求的记载范围内该效果均可实现,其中实施例1~2的实验数据(药物的释放时间)与实施例3的实验数据相比浮动不超过8%。
尽管已用具体实施例来说明和描述了本发明,然而应意识到,在不背离本发明的精神和范围的情况下可以作出许多其它的更改和修改。因此,这意味着在所附权利要求中包括属于本发明范围内的所有这些变化和修改。
- 还没有人留言评论。精彩留言会获得点赞!