人工湿地大颗粒基质表面微生物DNA的定性和/或定量提取方法与流程
本发明涉及微生物分子生态学技术领域,更具体地,涉及一种人工湿地砾石表面微生物DNA的定性和/或定量提取方法。
背景技术:
生物量和群落结构作为人工湿地的两大重要特性,它们在湿地内部的分布特征与污染物在湿地内部的降解行为息息相关。随着分子生物学的发展及其在生态学中的应用,特别是生态学方向宏基因组学方法的建立及发展,培养手段已远远不能满足对人工湿地大通量的物种信息需要。由此,研究便指向了包括培养及非培养微生物在内的整个环境基因组DNA上,通过高通量测序及qPCR等分子生物学技术手段,以这些更为准确的方式获得涵盖整个微生物群落的信息。而这些信息获得的前提,是需要提供足够质量、纯度和完整的基因组DNA,如华大基因有限公司的宏基因组测序送样要求为:总量≥1.5μg,OD260/280在1.8至2.0之间,OD260/280>1.5,且电泳检测条带无明显降解。当需要使用qPCR对细菌、古菌或氨氧化细菌等进行定量时,需要基因组DNA能够进行PCR反应,而且需要定量提取基因组DNA,数据重复性要好,以便准确定量基质中微生物的含量。
土壤、沉积物、淤泥等环境样品中含有大量的腐殖质,这是导致样品DNA提取效率及品质降低的主要因素之一,因此国内逐渐出现了各种可有效脱腐的DNA提取技术,以期从富含腐殖质的样品中有效的获得高品质的DNA。申请号为201010573017.8的中国专利申请公开了一种淡水沉积物总DNA的提取方法,申请号为201110090363.5的中国专利申请公开了一种土壤和沉积物总DNA提取方法及提取试剂盒,申请号为201110178774.X的中国专利申请公开了一种高效去除腐殖质的环境样品总DNA提取方法,申请号为201210137580.X的中国专利申请公开了一种从湖泊沉积物样品中提取纯化总DNA的高效方法,申请号为201410581883.X的中国专利申请公开了基于磁珠法提取土壤微生物DNA的试剂盒及方法等等。以上专利所公开的方法仅限于直接提取颗粒极细的环境样品,并未提及粗糙且表面不均一的大颗粒基质样品,尤其是如何对大颗粒基质样品表面微生物DNA进行定量的提取。人工湿地中为了防止系统堵塞,常会使用大颗粒砾石、火山石、沸石等基质作为床体填料,这些大颗粒基质的单粒重往往超过0.5g,无法像土壤、沉积物和淤泥那样使用微量土壤DNA提取试剂盒提取DNA,而且试剂盒价格昂贵,对于大批量样品的提取并非经济可行。
因此,需要研制适用于大颗粒基质表面微生物DNA提取的高效、快速、经济而且可批量化进行的方法,解决常规方法难以直接提取大颗粒基质样品表面微生物DNA、提取的DNA存在腐殖质等PCR抑制物以及定量不准确、DNA产量低的问题。
技术实现要素:
本发明的目的在于针对现有DNA提取技术难以直接获取大颗粒基质表面微生物DNA的不足之处,建立一种成本低、快速化并定量的提取人工湿地大颗粒基质表面微生物DNA的方法,不仅可以实现定性还可以定量检测分析。该方法所获得的DNA可直接作为PCR的模板,用于基因定量检测和高通量测序,开展人工湿地基质生物膜生物量和群落结构分析。
本发明目的通过以下技术方案予以实现:
提供一种人工湿地大颗粒基质表面微生物总DNA的定性和/或定量提取方法,该方法以人工湿地填充的大颗粒基质为材料,采用摇床震荡的方法洗脱砾石表面生物膜并离心获得沉淀,将沉淀用脱腐缓冲液和氯化钙溶液进行脱腐处理,再经SDS-溶菌酶法裂解细胞,氯仿抽提,异丙醇沉淀获得DNA。使用该方法获得的DNA,其OD260/280和OD260/280都在1.8左右。经本发明方法获得的DNA不需要稀释和纯化即可直接用于PCR反应。
所述大颗粒基质为材料包括砾石、火山石或沸石。
本发明方法所述提取方法包括基质生物膜洗脱和脱腐预处理和DNA抽提步骤。
具体地,其具体步骤如下:
S1.基质生物膜洗脱和脱腐预处理:
S11.采集大颗粒基质,加入无菌生理盐水,振荡洗脱大颗粒基质表面生物膜,得到悬浊液;
S12.吸取步骤S11所得悬浊液于适宜体积的无菌离心管中,离心处理后,弃净上清,继续加入悬浊液,再次离心处理后,待管内所得沉淀达到检测所设定的沉淀质量后,将含沉淀的离心管保存于-20℃冰箱备用;
S13.取出保存有沉淀的无菌离心管,在常温下放置解冻,将无菌石英砂加入沉淀中,向沉淀中加入脱腐缓冲液并涡旋混匀,离心处理后弃上清,所得沉淀进行下一步骤DNA提取操作。所述脱腐缓冲液的组成为100mmol/L Tris-HCl(pH8.0)、100mmol/L Na2P2O7、100mmol/L Na2EDTA(pH8.0),1%PVP,100mmol/L NaCl,0.05%Triton X-100;所述脱腐缓冲液的pH值为8.0。
S2.DNA提取:
S21.向步骤S13所得沉淀中加入0.5mol/L的氯化钙,涡旋混匀,离心处理后弃上清,得沉淀;
S22.向步骤S21所得沉淀中加入0.05mol/L的草酸钠,涡旋混匀,离心处理后弃上清,得沉淀;
S23.向步骤S22所得沉淀中加入DNA提取液和溶菌酶,涡旋混匀,摇床处理;
所述DNA提取液的组成为100mM Tris-HCl、1.5mol/L NaCl和1%CTAB,pH值为8.0;所述溶菌酶的浓度为100mg/mL;
S24.短暂离心后加入100uL 20%SDS,涡旋混匀,水浴处理,间隔颠倒摇匀数次;
S25.离心处理后收集中间液相层并重复洗涤沉淀一次;
S26.向收集的上清液体中加入等体积氯仿和异戊醇的混合液抽提,离心处理后收集上部的液相层;所述氯仿与异戊醇的混合体积比例为24:1;
S27.向收集的液相层中加入0.1倍体积的3mol/L乙酸钠和0.6倍体积的异丙醇,-20℃放20min;
S28.离心处理后弃上清;向沉淀中加入1mL70%乙醇洗涤,再次离心处理,弃上清,重复洗一次,得沉淀;
S29.将步骤S28中所得沉淀,烘干并溶解即获得DNA溶液。
优选地,步骤S11所述的生理盐水的浓度为0.85%。
优选地,步骤S11所述大颗粒基质和无菌生理盐水的质量体积比为1g:1mL。
优选地,步骤S11所述振荡洗脱是在无菌聚乙烯瓶中进行操作。
优选地,步骤S11所述振荡是于15℃、置于摇床后以225rpm的转速摇匀3h,可以充分地洗脱大颗粒基质(砾石)的表面生物膜。
优选地,步骤S12所述离心处理是在4℃、12000rp条件下离心处理10min。
优选地,步骤S13所述无菌石英砂的粒径约为1mm。
优选地,步骤S13所述离心处理是在室温、12000rp条件下离心处理5min。
优选地,步骤S13所述脱腐缓冲液按以下步骤制备:100mmol/L Tris-HCl(pH8.0)、100mmol/L Na2P2O7、100mmol/L Na2EDTA(pH8.0)、1%PVP、100mmol/L NaCl、0.05%Triton X-100、4%脱脂奶粉,pH8.0,用0.44μm纤维素膜抽滤,再用0.22μm无菌滤头抽滤。
优选地,步骤S21所述离心处理是在室温、12000rpm条件下离心5min。
优选地,步骤S22所述离心处理是在室温、12000rpm条件下离心5min。
优选地,步骤S23所述摇床处理的条件是在37℃、225rpm下处理30min;
优选地,步骤S24所述水浴处理的温度为65℃,水浴处理时间为1h。
优选地,步骤S25所述离心处理是在室温、12000rpm条件下离心5min。
优选地,步骤S26所述离心处理是在室温、12000rpm条件下离心10min。
优选地,步骤S26所述抽提的时间为10min。
优选地,步骤S27所述乙酸钠的pH值为5.2。
优选地,步骤S28所述离心处理是在室温、12000rpm条件下离心10min;所述再次离心处理是在室温、12000rpm条件下离心5min;
优选地,步骤S26所述收集中间液相层并重复洗涤一次按以下步骤进行:移液器吸取900μL中间液相层至一新离心管(2mL)后;沉淀重新加入800μL DNA提取液,100μL 20%SDS,涡旋混匀后65℃温浴10min,间隔颠倒摇匀数次,12000rpm室温离心5min,收集中间液相层800μL至一新离心管(2mL)。
优选地,步骤S29所述烘干并溶解DNA是按以下步骤进行:60℃烘干5min,加入50μL 10mmol/L的Tris-HCl(pH 8.0),手指轻弹溶解沉淀,将两管相同的样品DNA合并为100μL。
本发明所述方法所得DNA溶液,其OD260/280和OD260/280都在1.8左右,不需要稀释和纯化即可直接用于PCR反应,经电泳和质量计算,可实现定性和/或定量检测。
如果需要实现定量检测,在上述步骤的基础上,还包括以下步骤:
步骤S11还包括记录大颗粒基质的质量M和无菌生理盐水的体积V的步骤;在步骤S12包括记录所加悬浊液总体积V1步骤;计算沉淀所对应的大颗粒基质的质量M1为另称取30g所述相同的大颗粒基质,干燥后称重,质量记为m2。如此,m2/(30m1-30m0)即为沉淀的基质干重,从达到检测所设定的沉淀质量的管内沉淀中提取的总DNA标记为mgDNA。
计算公式:基质DNA含量(ng/g)=30mgDNA(m1-m0)/m2。
所述干燥后称重是将大颗粒基质置于烘箱70℃烘干48h。
所述提取方法与PCR和电泳方法结合应用于人工湿地大颗粒基质表面微生物总DNA的定性和/或定量检测方面。
本发明的有益效果:
本发明提供了一种人工湿地大颗粒基质表面微生物总DNA的定性和/或定量提取方法,本发明针对人工湿地填充的大颗粒基质,采用摇床震荡的方法成功洗脱砾石表面生物膜并离心获得沉淀,将沉淀用脱腐缓冲液和氯化钙溶液进行合理的脱腐处理,再经SDS-溶菌酶法裂解细胞,氯仿抽提,异丙醇沉淀获得DNA。使用该方法获得的DNA,其OD260/280和OD260/280都在1.8左右。经本发明方法获得的DNA不需要稀释和纯化即可直接用于PCR反应。建立一种成本低、快速化并定量的提取人工湿地大颗粒基质表面微生物DNA的方法,该方法所获得的DNA可直接作为PCR的模板,不需要稀释和纯化,可很好地用于基因定量检测和高通量测序,开展人工湿地基质生物膜生物量和群落结构分析。
基于本发明方法,通过分步记录相关质量,可以成功定量检测人工湿地大颗粒基质表面微生物总DNA。
附图说明
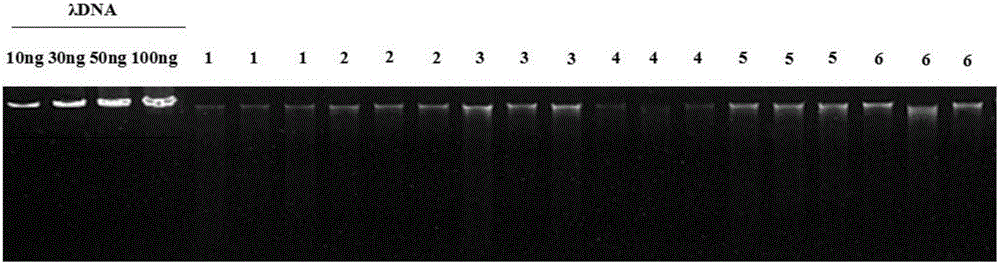
图1为未经脱腐处理直接提取的DNA电泳检测图。图中编号1~6依次为S-HF+UP、B-HF+UP、S-HF+TD、B-HF+TD、S-HF+CI、B-HF+CI,点样量均为2uL。λDNA作为定量标准(北京鼎国昌盛生物技术有限公司)上样量从左至右依次为10ng、30ng和50ng。
图2为试剂盒法提取的DNA电泳检测图。图中编号1~6样品与图1相同,点样量均为2uL,各提取三次作为平行。λDNA作为定量标准上样量从左至右依次为10ng、30ng、50ng和100ng。
图3为本发明的方法提取的DNA电泳检测图。图中编号1~6样品及上样量与图1相同。λDNA作为定量标准上样量从左至右依次为10ng、30ng、50ng和100ng。
图4为以本发明的方法提取的DNA直接作为模板扩增16S rRNA基因全长电泳检测图。图中编号0表示模板为10mmol/L的Tris-HCl(pH 8.0),编号1~6依次表示模板来自S-HF+UP、B-HF+UP、S-HF+TD、B-HF+TD、S-HF+CI、B-HF+CI,PCR上样量为2uL,重复编号为同一样品提取三次。M为DNA分子量标准(TaKaRa,Dalian,China)从上至下依次为2000bp、1000bp、750bp、500bp、250bp、100bp(bp:碱基对)。
图5为以试剂盒法提取的DNA直接作为模板扩增16S rRNA基因全长电泳检测图。图中编号、上样量及DNA分子量标准与图4相同。
图6为荧光定量PCR检测的细菌16S rRNA基因拷贝数。
图7为以试剂盒法和本发明的方法提取的DNA为模板扩增16S rRNAV3区的DGGE检测图,左边编号1~6依次表示采用某试剂盒方法,其模板来自S-HF+UP、B-HF+UP、S-HF+TD、B-HF+TD、S-HF+CI、B-HF+CI,DGGE上样量为2μg;右边编号1~6依次表示采用本发明方法,其模板来自S-HF+UP、B-HF+UP、S-HF+TD、B-HF+TD、S-HF+CI、B-HF+CI,DGGE上样量为2μg。
具体实施方式
下面结合附图和实施过程对本发明作进一步说明,除非特别说明,本发明采用的试剂及仪器为本技术领域常规的试剂及仪器。
实施例1:从人工湿地砾石表面提取微生物DNA
人工湿地砾石样品采集:
本实施例从一组处理生活污水的水平潜流人工湿地采集砾石样品,湿地装置长80cm、宽60cm、高80cm。湿地基质为砾石(粒径1~2cm),填充高度为60cm,分别设置无植物组,美人蕉组和再力花组。装置水力负荷为2.5m/d,全天连续运行,分别于近进水端及近出水端采集基质样品,表层0~10cm处基质混合为表层样品,底层30~40cm处基质混合为底层样品,三个装置共6个样品,其中3个为表层样品,3个为底层样品,各采集约200g基质装入无菌密封袋备用,各样品分别命名为:S-HF+UP(水平潜流无植物组表层基质)、B-HF+UP(水平潜流无植物组底层基质)、S-HF+TD(水平潜流再力花组表层样品)、B-HF+TD(水平潜流再力花组底层样品)、S-HF+CI(水平潜流美人蕉组表层样品)、B-HF+CI(水平潜流美人蕉组底层样品)。
基于前述采集的样品,本实施例提供一种人工湿地(用于处理城镇居民每日排放的污水)砾石表面微生物DNA的定量提取方法,包括以下步骤:
S1.样品预处理
S11.采集人工湿地砾石装入无菌密封袋,将密封袋中砾石缓慢倒入500mL的无菌聚乙烯瓶,至砾石重量为100g为止,再往瓶中加入100mL无菌生理盐水(0.85%),置于摇床15℃以225rpm的转速摇匀3h,以充分洗脱砾石表面生物膜。
S12.吸取悬浊液于适宜体积的无菌离心管中,12000rpm 4℃离心10min,弃净上清,继续加入悬浊液,同上离心后,管内所得沉淀达到检测所设定的沉淀质量后(0.5g),将含沉淀离心管保存于-20℃冰箱备用。
S13.取出保存有0.5g沉淀的离心管,在常温下放置3min解冻,将0.4g无菌石英砂(粒径约1mm)加入沉淀中,向沉淀中加入1.5mL脱腐缓冲液[100mmol/LTris-HCl(pH8.0),100mmol/L Na2P2O7,100mmol/L Na2EDTA(pH8.0),1%PVP,100mmol/L NaCl,0.05%Triton X-100,pH 8.0],涡旋混匀,12000rpm室温离心5min,弃上清,得沉淀备DNA提取用。
S2.DNA提取
S21.向沉淀中加入1mL 0.5mol/L氯化钙,涡旋混匀,12000rpm室温离心5min,弃上清。
S22.向沉淀中加入1mL 0.05mol/L草酸钠,涡旋混匀,12000rpm室温离心5min,弃上清。
S23.向沉淀中加入800μL DNA提取液(100mM Tris-HCl,1.5mol/L NaCl,1%CTAB,pH8.0)和100μL溶菌酶(100mg/mL),涡旋混匀,置于摇床37℃225rpm30min。
S24.短暂离心,加入100uL 20%SDS,涡旋混匀,65℃水浴1h,间隔颠倒摇匀数次。
S25.12000rpm室温离心5min,收集中间液相层900μL至一新2mL离心管;沉淀加入800μL DNA提取液,90μL 20%SDS,涡旋混匀后65℃温浴10min,间隔颠倒摇匀数次,同上离心,收集中间液相层800μL至一新2mL离心管。
S26.向收集的上清液体中加入等体积氯仿:异戊醇(24:1)抽提10min,然后12000rpm离心10min,收集上部液相层。
S27.向收集液相层中加入0.1倍体积的3mol/L乙酸钠(pH5.2)和0.6倍体积的异丙醇,-20℃放20min。
S28.12000rpm离心10min,弃上清;向沉淀中加入1mL70%乙醇洗涤,12000rpm离心5min,弃上清,重复洗一次。
S29.60℃烘干5min,加入50μL 10mmol/L的Tris-HCl(pH 8.0),手指轻弹溶解沉淀,将两管相同样品合并成100μL。
使用16S rDNA引物27F(5’AGAGTTTGATCCTGGCTCAG3’)和1492R(5’GGTTACCTTGTTACGACTT3’),检测gDNA的16S rDNA基因全长PCR扩增效果。PCR扩增体系具体组成如下:5μL 10×buffer,5μL dNTP(2mmol/L),1.5μL正向引物27F(10μmol/L),1.5μL反向引1492R(10μmol/L),0.5μLTaq DNA pol-ymerase(TaKaRa,Dalian,China),4μL MgCl2(25mmol/L),1μL的DNA模板,加灭菌的双蒸水至50μL。用PCR热循环仪(Bio-rad S1000,US)进行热循环,具体热循环条件如下:采用降落PCR扩增,94℃预变性5min,94℃变性1min,63.5℃退火1min,每个循环退火温度降1℃,72℃延伸3min,共10个循环,94℃变性1min,53.5℃退火1min,72℃延伸3min,共20个循环,72℃延伸15min。PCR反应产物2μL用2%琼脂糖凝胶在1×TAE缓冲液中电泳检测。
电泳方法:基因组DNA的琼脂糖凝胶电泳取2μL DNA溶解液与0.5μL 6×上样缓冲液混匀,点样于1%琼脂糖凝胶(含1×Gelred)上,置于TAE缓冲液(pH8.0)进行电泳95V 18min后,使用凝胶成像系统(Bio-rad,US)拍照分析。
表1本实施例提取基质微生物DNA纯度
实施例2:从人工湿地砾石表面提取微生物DNA
人工湿地砾石样品采集:
本实施例从一组处理生活污水的水平潜流人工湿地采集砾石样品,湿地装置长80cm、宽60cm、高80cm,湿地基质为砾石(粒径1~2cm),填充高度为60cm,分别设置无植物组,美人蕉组和再力花组。装置水力负荷为2.5m/d,全天连续运行,分别于近进水端及近出水端采集基质样品,表层0~10cm处基质混合为表层样品,底层30~40cm处基质混合为底层样品,三个装置共6个样品,其中3个为表层样品,3个为底层样品,各采集约200g基质装入无菌密封袋备用,各样品分别命名为:S-HF+UP(水平潜流无植物组表层基质)、B-HF+UP(水平潜流无植物组底层基质)、S-HF+TD(水平潜流再力花组表层样品)、B-HF+TD(水平潜流再力花组底层样品)、S-HF+CI(水平潜流美人蕉组表层样品)、B-HF+CI(水平潜流美人蕉组底层样品)。
基于前述采集的样品,本实施例提供一种人工湿地(用于处理城镇居民每日排放的污水)砾石表面微生物DNA的定量提取方法,包括以下步骤:
S1.样品预处理
S11.采集人工湿地砾石装入无菌密封袋,将密封袋中砾石缓慢倒入500mL的无菌聚乙烯瓶,至砾石重量为100g为止,再往瓶中加入100mL无菌生理盐水(0.85%),置于摇床15℃以225rpm的转速摇匀3h,以充分洗脱砾石表面生物膜。
S12.吸取悬浊液2mL于2mL无菌离心管中(空管称重记为m0),12000rpm 4℃离心10min,弃净上清,将含有沉淀的管称重并记为m1,沉淀质量即为m1-m0,继续加入悬浊液体积为1/(m1-m0)-2,同上离心后,管内即得0.5g沉淀,相当于1/(m1-m0)的砾石加入量,将含沉淀离心管保存于-20℃冰箱备用。
另称取30g砾石,置于烘箱70℃烘干48h,干燥后的砾石称重,重量记为m2。如此,m2/(30m1-30m0)即为0.5g沉淀的基质干重,从该0.5g沉淀中提取的总DNA标记为mgDNA。计算公式:基质DNA含量(ng/g)=30mgDNA(m1-m0)/m2。
S13.取出保存有0.5g沉淀的离心管,在常温下放置3min解冻,将0.4g无菌石英砂(粒径约1mm)加入沉淀中,向沉淀中加入1.5mL脱腐缓冲液[100mmol/LTris-HCl(pH8.0),100mmol/L Na2P2O7,100mmol/L Na2EDTA(pH8.0),1%PVP,100mmol/L NaCl,0.05%Triton X-100,pH 8.0],涡旋混匀,12000rpm室温离心5min,弃上清,得沉淀备DNA提取用。
S3.DNA提取
S21.向沉淀中加入1mL 0.5mol/L氯化钙,涡旋混匀,12000rpm室温离心5min,弃上清。
S22.向沉淀中加入1mL 0.05mol/L草酸钠,涡旋混匀,12000rpm室温离心5min,弃上清。
S23.向沉淀中加入800μL DNA提取液(100mM Tris-HCl,1.5mol/L NaCl,1%CTAB,pH8.0)和100μL溶菌酶(100mg/mL),涡旋混匀,置于摇床37℃225rpm30min。
S24.短暂离心,加入100uL 20%SDS,涡旋混匀,65℃水浴1h,间隔颠倒摇匀数次。
S25.12000rpm室温离心5min,收集中间液相层900μL至一新2mL离心管;沉淀加入800μL DNA提取液,90μL 20%SDS,涡旋混匀后65℃温浴10min,间隔颠倒摇匀数次,同上离心,收集中间液相层800μL至一新2mL离心管。
S26.向收集的上清液体中加入等体积氯仿:异戊醇(24:1)抽提10min,然后12000rpm离心10min,收集上部液相层。
S27.向收集液相层中加入0.1倍体积的3mol/L乙酸钠(pH5.2)和0.6倍体积的异丙醇,-20℃放20min。
S28.12000rpm离心10min,弃上清;向沉淀中加入1mL70%乙醇洗涤,12000rpm离心5min,弃上清,重复洗一次。
S29.60℃烘干5min,加入50μL 10mmol/L的Tris-HCl(pH 8.0),手指轻弹溶解沉淀,将两管相同样品合并成100μL。
使用16S rDNA引物27F(5’AGAGTTTGATCCTGGCTCAG3’)和1492R(5’GGTTACCTTGTTACGACTT3’),检测gDNA的16S rDNA基因全长PCR扩增效果。PCR扩增体系具体组成如下:5μL 10×buffer,5μL dNTP(2mmol/L),1.5μL正向引物27F(10μmol/L),1.5μL反向引1492R(10μmol/L),0.5μLTaq DNA pol-ymerase(TaKaRa,Dalian,China),4μL MgCl2(25mmol/L),1μL的DNA模板,加灭菌的双蒸水至50μL。用PCR热循环仪(Bio-rad S1000,US)进行热循环,具体热循环条件如下:采用降落PCR扩增,94℃预变性5min,94℃变性1min,63.5℃退火1min,每个循环退火温度降1℃,72℃延伸3min,共10个循环,94℃变性1min,53.5℃退火1min,72℃延伸3min,共20个循环,72℃延伸15min。PCR反应产物2μL用2%琼脂糖凝胶在1×TAE缓冲液中电泳检测。
电泳方法:基因组DNA的琼脂糖凝胶电泳取2μL DNA溶解液与0.5μL 6×上样缓冲液混匀,点样于1%琼脂糖凝胶(含1×Gelred)上,置于TAE缓冲液(pH8.0)进行电泳95V 18min后,使用凝胶成像系统(Bio-rad,US)拍照分析。
表2本实施例提取基质微生物DNA获得量及纯度
实施例3对比试验
为了比较本发明(实施例1和实施例2,以下数据和样品中称本发明)与现有的未脱腐处理法和试剂盒法的提取效果,本申请人同时进行了利用未脱腐处理法和试剂盒法的实验,具体操作如下:
(1)该方法未进行氯化钙和草酸钠处理,其他步骤与实施例1或实施例2相同,DNA抽提液的配方为(100mM Tris-HCl,100mM EDTA,200mM NaCl,2%PVPP,3%CTAB,pH8.0)。
(2)使用 DNA Isolation Kit(MO BIO Laboratories Inc.,Carlsbad,CA,USA)提取基因组DNA,将PowerBead Tubes中的液体及研磨珠全部转移至装有0.5g沉淀的2mL离心管中,其余步骤根据试剂盒步骤进行操作。
上述两种方法使用的基质样品与本发明相同,即样品处理都用摇床洗脱并离心为0.5g沉淀,提取的DNA都溶于100μL 10mmol/L的Tris-HCl(pH 8.0)中。
DNA浓度和纯度检测
(1)基因组DNA的琼脂糖凝胶电泳取2μL DNA溶解液与0.5μL 6×上样缓冲液混匀,点样于1%琼脂糖凝胶(含1×Gelred)上,置于TAE缓冲液(pH8.0)进行电泳95V 18min后,使用凝胶成像系统(Bio-rad,US)拍照分析。
按照实施例1或实施例2中步骤S1的方法(图3)和试剂盒法(图2)提取的DNA均具有较高的完整性,而且无明显的RNA条带,电泳定量的结果也显示,本发明所提取的DNA量要高于试剂盒法;未经脱腐处理(图1)提取的DNA量少条带较弱,有明显的降解,而且有较强的RNA条带。这表明本发明的提取方法具有较高的DNA获得率,且DNA具有较高完整性,脱腐对杂质有较好的去除效果。
(2)超微量分光光度计测定DNA产量与纯度直接取1μL提取好的gDNA原液,置于Nanodrop ND-2000超微量分光光度计(NanoDrop Technologies,Wilmington,DE,USA)上,测定其浓度及OD260/280和OD230/260。
按照实施例1中S1的方法提取获得的DNA,如表1所示,其单次提取DNA总量皆超过1.5μg,OD260/280和OD260/230都在1.8左右,结果较为稳定,无论在总量还是品质上,都能满足宏基因组测序的送样要求;而试剂盒方法提取获得的DNA,单次提取DNA总量只有部分样品能达到宏基因组测序的送样要求,与本发明相比稳定性较差,且试剂盒单次提取DNA总量极显著小于本发明(p<0.01);这说明本发明提取的人工湿地大颗粒基质表面微生物DNA质量和品质都优于 DNA Isolation Kit。
表3本发明与试剂盒法提取基质微生物DNA获得量及纯度结果比较
PCR检测:
(1)使用16S rDNA引物27F(5’AGAGTTTGATCCTGGCTCAG3’)和1492R(5’GGTTACCTTGTTACGACTT3’),检测gDNA的16S rDNA基因全长PCR扩增效果。PCR扩增体系具体组成如下:5μL 10×buffer,5μL dNTP(2mmol/L),1.5μL正向引物27F(10μmol/L),1.5μL反向引1492R(10μmol/L),0.5μLTaq DNA pol-ymerase(TaKaRa,Dalian,China),4μL MgCl2(25mmol/L),1μL的DNA模板,加灭菌的双蒸水至50μL。用PCR热循环仪(Bio-rad S1000,US)进行热循环,具体热循环条件如下:采用降落PCR扩增,94℃预变性5min,94℃变性1min,63.5℃退火1min,每个循环退火温度降1℃,72℃延伸3min,共10个循环,94℃变性1min,53.5℃退火1min,72℃延伸3min,共20个循环,72℃延伸15min。PCR反应产物2μL用2%琼脂糖凝胶在1×TAE缓冲液中电泳检测。
按照实施例1或实施例2的方法和试剂盒法提取获得的DNA,直接作为模板扩增16S rRNA基因全长。如图4和5,对照为10mmol/L的Tris-HCl(pH 8.0)溶液均未扩增出目的大小的条带;试剂盒法提取的样品中,标记为6的其中一个样品未能扩增出目的大小的条带;但本发明的方法提取的全部样品都能扩出大小约1500bp的目的条带,而且没有明显的引物二聚体。因此,通过本发明的方法能获得纯度较高的DNA,直接用于PCR扩增及细菌16S rRNA基因信息分析。
(2)使用引物341F(5’CCTACGGGAGGCAGCAG3’)和534R(5’ATTACCGCGGCTGCTGG3’),实时定量PCR测定基质的细菌16S rRNA基因拷贝数。实时定量PCR体系具体组成如下:10μL SYBR Premix Ex TaqTM(Takara,Japan),引物(10μM)各0.4μL,2μL模板DNA,无菌去离子水补齐至20μL。使用实时定量PCR仪(Bio-radCFX96,US)测定,具体扩增程序为,95℃预变性30s;40个循环如下过程:95℃变性15s,62℃退火20s,72℃延伸30s。
按照实施例1或实施例2中S1的方法和试剂盒方法提取获得的DNA,直接作为模板进行实时定量PCR,结果显示本发明的方法测得的细菌16S rRNA基因拷贝数极显著高于试剂盒法(p<0.01,图6),这表明本发明的提取方法与试剂盒法相比,细胞的裂解程度更高,更能反映样品的全部细菌生物量,能更准确和全面的描述人工湿地系统中的细菌生物量。
(3)PCR扩增16S rRNAV3区
使用引物与实时定量PCR相同,但需要在341F的5’端添加GC夹子5’CGCCCGCCGCGCGCGGCGGGCGGGGCGGGGGC ACGGGGGG3’,进行变性梯度凝胶电泳(DGGE)检测基质的细菌群落结构组成。PCR扩增体系具体组成如下:5μL10×buffer,5μL dNTP(2mmol/L),1.5μL正向引物27F(10μmol/L),1.5μL反向引1492R(10μmol/L),0.5μL Taq DNA polymerase(TaKaRa,Dalian,China),4μLMgCl2(25mmol/L),1μL的DNA模板,加灭菌的双蒸水至50μL。用PCR热循环仪(Bio-rad S1000,US)进行热循环,具体热循环条件如下:采用降落PCR扩增,94℃预变性10min,94℃变性30s,65℃退火30s,每个循环退火温度降0.5℃,72℃延伸1min,共19个循环,94℃变性30s,55℃退火30s,72℃延伸1min,共14个循环,72℃延伸10min。PCR反应产物2μL用2%琼脂糖凝胶在1×TAE缓冲液中电泳检测,并使用定量DNA对每个PCR产物样品的目的条带进行定量。各PCR产物加入10μL 10×Loading Buffer(TaKaRa,Dalian,China)并混匀,置于烘箱中60℃过夜浓缩,根据电泳定量结果,向各个产物样品加入无菌去离子水调节条带浓度至100ng/μL,DGGE上样量为20μL。采用DcodeTM基因突变检测系统(Bio-rad,US)对PCR反应产物进行分离,制备8%的聚丙烯酰胺凝胶,变性剂浓度范围为40~60%(100%的变性剂中含有7mol/L的尿素和40%40的去离子甲酰胺)。
电泳条件为:60℃及110V电压下,电泳12h。电泳结果经Gel Red核酸染料染色后用凝胶成像系统(Bio-rad,US)观察并用Quantity One软件进行分析。样品的丰富度(S)用条带数量表示,香浓-威纳指数(H')计算公式为:H'=-∑(pi)(lnpi)。其中pi值是每条DNA条带亮度密度与每个电泳泳道中DNA条带亮度总和的比值;lnpi是pi的自然对数。
按照实施例1或实施例2中方法和试剂盒方法提取获得的DNA,直接作为模板扩增细菌16S rRNA V3区后进行DGGE分析,如图7显示与试剂盒法相比,本发明的样品条带更清晰且条带之间能明显隔开。由表2可知,本发明所得的细菌丰富度和多样性均高于试剂盒法,说明本发明提取的DNA含有的微生物多样性信息量要高于试剂盒法,能更有效的反映人工湿地基质微生物群落的多样性。
表4本发明与试剂盒法的细菌丰富度和多样性结果
- 还没有人留言评论。精彩留言会获得点赞!