芳胺氮芥衍生物N,N‑二(2‑氯乙基)‑N′‑乙酰基‑1,4‑苯二胺及其制备方法与流程
本发明涉及氮芥类抗肿瘤药物的技术领域,具体涉及芳胺氮芥衍生物N,N-二(2-氯乙基)-N′-乙酰基-1,4-苯二胺及其制备方法。
背景技术:
氮芥类药物是常用的一种抗肿瘤药物,具有特别好的抗肿瘤活性,但是作为传统的细胞毒类药物,其仍具有生物烷化剂所特有的缺点,在抑制和毒害增生活跃的肿瘤细胞的同时,对其它增生较快的正常细胞也产生抑制作用,例如对一些骨髓细胞、肠上皮细胞、还有一些毛发细胞也具有阻扰灭杀的效果,这就会引起例如恶心、呕吐、脱发等严重的不良反应;另外,氮芥类药物还具有免疫抑制作用,会降低机体的防御功能,使细菌病毒等趁虚而入,引起各种感染。
氮芥类抗肿瘤药物从构造上看由两部分组成:烷基化部分和载体部分,烷基化部分是主体部分,也是抗肿瘤活性的功能基团,载体部分可看成结构修饰的部分,对其进行结构修饰可以不断提高药物在人体内的吸收、分布,进而有助于提高对肿瘤细胞的选择性,同时也能有效的抑制副作用的产生。
因此,如何对氮芥衍生物进行修饰,合成出具有较低的毒副作用的氮芥,已成为目前研究的热点。
技术实现要素:
针对现有技术中存在的问题,本发明的目的在于提供一种具有较低毒副作用的且能增强肿瘤治疗效果,同时兼具杀菌消炎疗效的芳胺氮芥衍生物N,N-二(2-氯乙基)-N′-乙酰基-1,4-苯二胺,其可达到降低病人化疗后因免疫能力下降而引起并发症的风险。
本发明的另一目的为提供上述芳胺氮芥衍生物N,N-二(2-氯乙基)-N′-乙酰基-1,4-苯二胺的制备方法。
为了达到上述目的,本发明采用以下技术方案予以实现。
芳胺氮芥衍生物N,N-二(2-氯乙基)-N′-乙酰基-1,4-苯二胺,其特征在于,结构如式Ⅰ:
上述芳胺氮芥衍生物N,N-二(2-氯乙基)-N′-乙酰基-1,4-苯二胺的制备方法,其特征在于,包括以下步骤:
步骤1,制备N,N-二(2-氯乙基)-1,4-苯二胺;
步骤2,向反应器中加入反应原料N,N-二(2-氯乙基)-1,4-苯二胺、二氯甲烷和三乙胺,冰水浴冷却,搅拌,并向反应器中滴加乙酰氯和二氯甲烷的混合溶液,滴加完成后,撤掉冰水浴,室温反应2-14小时,待反应完全后,使用蒸馏水洗涤反应液,且用无水硫酸铜干燥,常压蒸馏,对蒸馏后的滤饼进行纯化,即得。
步骤1包含以下子步骤:
子步骤1a,首先按照以下步骤制备N,N-二(2-羟乙基)-4-硝基苯胺:将4-氯硝基苯、二乙醇胺、碳酸钾和质量百分比为1%的CuSO4溶液依次加入反应器中,搅拌均匀,升温至82℃时,向反应器中加入甲苯,继续升温至115-120℃,恒温搅拌反应6-9小时,待反应完全后,蒸馏去除甲苯,将蒸馏后的母液倒入40-60℃的热水中,搅拌,静置过夜后抽滤水层得粗品,使用无水乙醇对所述粗品重结晶,得黄色针状晶体N,N-二(2-羟乙基)-4-硝基苯胺。
优选地,子步骤1a中,所述4-氯硝基苯、二乙醇胺、碳酸钾的摩尔比为(1.00∶1.20∶0.60)-(1.00∶1.40∶0.7)。
优选地,子步骤1a中,所述质量百分比为1%的CuSO4溶液与所述4-氯硝基苯的用量的比例为(0.2-0.5)mL:(2-3)g。
优选地,子步骤1a中,所述甲苯与所述4-氯硝基苯的用量的比例为1mL:(2-3)g。
子步骤1b,然后按照以下步骤制备N,N-二(2-氯乙基)-4-硝基苯胺:取所述黄色针状晶体N,N-二(2-羟乙基)-4-硝基苯胺溶于装有干燥的二氯甲烷的反应器中,并向所述反应器中加入三乙胺;再在室温下搅拌滴加二氯亚砜和二氯甲烷的混合溶液,所述滴加时间为1-1.5小时,升高温度至40-45℃,回流,同时搅拌反应2小时,反应完全后静置冷却,并用饱和碳酸氢钠溶液洗涤,合并有机层并用无水硫酸钠干燥,减压蒸馏去除溶剂,得N,N-二(2-氯乙基)-4-硝基苯胺的粗品,使用无水乙醇对所述粗品重结晶,得黄色针状晶体N,N-二(2-氯乙基)-4-硝基苯胺。
优选地,子步骤1b中,所述黄色针状晶体N,N-二(2-羟乙基)-4-硝基苯胺与所述二氯亚砜的摩尔比为1:1-1:2。
优选地,子步骤1b中,所述黄色针状晶体N,N-二(2-羟乙基)-4-硝基苯胺与所述二氯甲烷、所述三乙胺的用量比例为2.7g:60mL:1.1mL。
优选地,子步骤1b中,所述二氯亚砜与所述二氯甲烷的体积比为1:(13-15)。
子步骤1c,最后按照以下步骤制备N,N-二(2-氯乙基)-1,4-苯二胺:取所述黄色针状晶体N,N-二(2-氯乙基)-4-硝基苯胺溶于装有浓盐酸的反应器中,并向所述反应器中加入氯化亚锡,在氮气氛围下回流搅拌反应,待颜色变为无色透明时停止反应;使用0℃的冰水浴冷却,并用浓氨水调pH至7.5~8.0,用乙酸乙酯萃取,无水硫酸钠干燥,减压蒸馏溶剂,即得N,N-二(2-氯乙基)-1,4-苯二胺。
优选地,子步骤1c中,所述黄色针状晶体N,N-二(2-氯乙基)-4-硝基苯胺与所述氯化亚锡、所述浓盐酸的用量比为1mmol:(1-6)mol:(10-50)mL。
优选地,步骤2中,所述反应原料中,N,N-二(2-氯乙基)-1,4-苯二胺与所述二氯甲烷的用量比为1.00g:20.0mL;所述N,N-二(2-氯乙基)-1,4-苯二胺与所述三乙胺的摩尔比为1:3-1:5。
优选地,步骤2中,所述N,N-二(2-氯乙基)-1,4-苯二胺与所述乙酰氯的摩尔比为1:1.5-1:2.5;所述乙酰氯与所述二氯甲烷的混合溶液中,所述乙酰氯与所述二氯甲烷的摩尔比为1:3-1:5。
优选地,步骤2中,所述滴加的时间为1-2小时。
优选地,步骤2中,所述纯化采用柱层析技术,所述柱层析技术中的流动相为石油醚与乙酸乙酯的混合物,且所述石油醚与所述乙酸乙酯的体积比为1:1-1:3。
与现有技术相比,本发明的有益效果为:
本发明以N,N-二(2-氯乙基)-1,4-苯二胺为药效基团,以乙酰氯对N,N-二(2-氯乙基)-1,4-苯二胺进行结构修饰,使得本发明的芳胺氮芥衍生物N,N-二(2-氯乙基)-N′-乙酰基-1,4-苯二胺在具有增强氮芥治疗指数的前提下,又能有效降低氮芥的毒副作用,同时兼具杀菌、消炎的疗效,以达到降低病人化疗后因免疫力下降而引起的并发症的风险。
附图说明
下面结合附图和具体实施例对本发明做进一步详细说明。
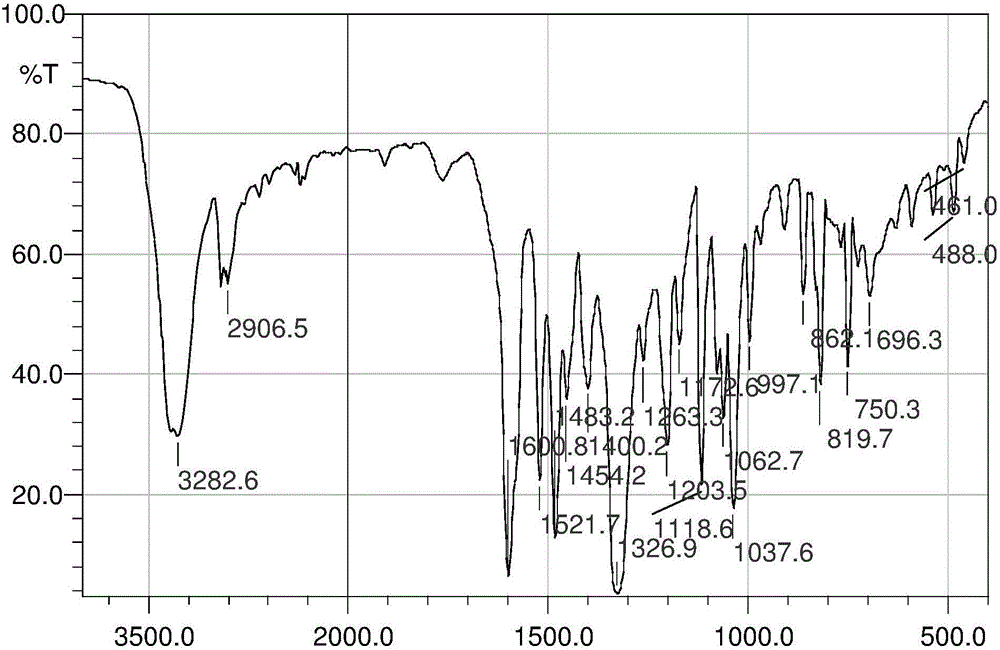
图1为N,N-二(2-羟乙基)-4-硝基苯胺的红外光谱图;图中,横坐标为波数,单位为cm-1,纵坐标为透光率,单位为%;
图2为N,N-二(2-氯乙基)-4-硝基苯胺的红外光谱图;图中,横坐标为波数,单位为cm-1,纵坐标为透光率,单位为%;
图3为N,N-二(2-氯乙基)-1,4-苯二胺的红外光谱图;图中,横坐标为波数,单位为cm-1,纵坐标为透光率,单位为%;
图4为芳胺氮芥衍生物N,N-二(2-氯乙基)-N′-乙酰基-1,4-苯二胺的红外光谱图;图中,横坐标为波数,单位为cm-1,纵坐标为透光率,单位为%;
图5为芳胺氮芥衍生物N,N-二(2-氯乙基)-N′-乙酰基-1,4-苯二胺的核磁谱图;图中,横坐标是化学位移,单位为ppm。
具体实施方式
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域的技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限制本发明的范围。
本发明提供一种具有较低毒副作用的且能增强肿瘤治疗效果,同时兼具杀菌消炎疗效的芳胺氮芥衍生物N,N-二(2-氯乙基)-N′-乙酰基-1,4-苯二胺,其结构如式Ⅰ:
芳胺氮芥衍生物N,N-二(2-氯乙基)-N′-乙酰基-1,4-苯二胺的制备方法,包括以下步骤:
步骤1,制备N,N-二(2-氯乙基)-1,4-苯二胺;
子步骤1a,首先按照以下步骤制备N,N-二(2-羟乙基)-4-硝基苯胺:将4-氯硝基苯、二乙醇胺、碳酸钾和质量百分比为1%的CuSO4溶液依次加入反应器中,搅拌均匀,升温至82℃时,向反应器中加入甲苯,继续升温至115-120℃,恒温搅拌反应6-9小时,待反应完全后,蒸馏去除甲苯,将蒸馏后的母液倒入40℃-60℃的热水中,搅拌,静置过夜后抽滤水层得粗品,使用无水乙醇对所述粗品重结晶,得黄色针状晶体N,N-二(2-羟乙基)-4-硝基苯胺。
子步骤1b,然后按照以下步骤制备N,N-二(2-氯乙基)-4-硝基苯胺:取所述黄色针状晶体N,N-二(2-羟乙基)-4-硝基苯胺溶于装有干燥的二氯甲烷的反应器中,并向所述反应器中加入三乙胺,其中,所述黄色针状晶体N,N-二(2-羟乙基)-4-硝基苯胺与所述二氯甲烷、所述三乙胺的用量比例为2.7g:60mL:1.1mL;再在室温下搅拌滴加二氯亚砜和二氯甲烷的混合溶液,所述滴加时间为1小时,升高温度至40℃,回流,同时搅拌反应2小时,反应完全后静置冷却,并用饱和碳酸氢钠溶液洗涤,合并有机层并用无水硫酸钠干燥,减压蒸馏去除溶剂,得N,N-二(2-氯乙基)-4-硝基苯胺的粗品,使用无水乙醇对所述粗品重结晶,得黄色针状晶体N,N-二(2-氯乙基)-4-硝基苯胺。
子步骤1c,最后按照以下步骤制备N,N-二(2-氯乙基)-1,4-苯二胺:取所述黄色针状晶体N,N-二(2-氯乙基)-4-硝基苯胺溶于装有浓盐酸的反应器中,并向所述反应器中加入氯化亚锡,在氮气氛围下回流搅拌反应,待颜色变为无色透明时停止反应;使用0℃的冰水浴冷却,并用浓氨水调pH至7.5~8.0,用乙酸乙酯萃取,无水硫酸钠干燥,减压蒸馏溶剂,即得。
步骤2,向反应器中加入反应原料N,N-二(2-氯乙基)-1,4-苯二胺、二氯甲烷N,N-二(2-羟乙基)-4-硝基苯胺和三乙胺,冰水浴冷却,搅拌,并向反应器中滴加乙酰氯和二氯甲烷N,N-二(2-氯乙基)-4-硝基苯胺的混合溶液,滴加的时间为1-2小时,滴加完成后,撤掉冰水浴,在室温下反应至少2-14小时,待反应完全后,使用蒸馏水洗涤反应液,且用无水硫酸铜干燥,常压蒸馏,对蒸馏后的滤饼进行纯化,即得。
实施例1
1.1 N,N-二(2-羟乙基)-4-硝基苯胺的合成
向250mL三口烧瓶内分别加入4-氯硝基苯12.64g(0.080mol)、二乙醇胺10.99g(0.104mol)和7.18g(0.052mol)碳酸钾,最后加入1.2mL质量浓度为1%的CuSO4溶液,混合均匀。加热升高温度,当温度达到82℃时,向反应液中加入5.0mL的甲苯,继续升高温度控制反应温度为117℃,使用薄层色谱法跟踪反应进程,恒温搅拌反应6h。待反应完全后,蒸馏去除甲苯,将蒸馏后的母液倒入40℃的热水中,剧烈搅拌,静置过夜后抽滤除去水层得粗品,粗品用无水乙醇重结晶,得黄色针状晶体N,N-二(2-羟乙基)-4-硝基苯胺11.39g,收率63.0%。
1.2 N,N-二(2-氯乙基)-4-硝基苯胺的合成
称取2.70g(0.012mol)黄色针状晶体N,N-二(2-羟乙基)-4-硝基苯胺即化合物N,N-二(2-羟基乙基)-4-硝基苯胺,溶于60.0mL干燥的二氯甲烷中,然后向反应瓶中加入1.1mL的三乙胺;取1.3mL(0.018mol)二氯亚砜溶于20.0mL二氯甲烷溶液,室温搅拌下将该溶液滴加到反应瓶,滴加的时间为lh,滴加完成后,加热至40℃,回流,并在回流的状态下搅拌反应2h。反应完全后静置冷却,用30.0mL干燥的二氯甲烷稀释,并分别用20.0mL的饱和碳酸氢钠溶液洗涤三次,合并有机层用无水硫酸钠干燥,0.01MPN,N-二(2-羟乙基)-4-硝基苯胺下减压蒸馏除去溶剂,得N,N-二(2-氯乙基)-4-硝基苯胺的粗品。粗品用无水乙醇重结晶得黄色针状晶体N,N-二(2-氯乙基)-4-硝基苯胺2.99g,收率95.0%。
1.3 N,N-二(2-氯乙基)-1,4-苯二胺的合成
称取1.04g(4mmol)黄色针状晶体N,N-二(2-氯乙基)-4-硝基苯胺溶于40.0mL浓盐酸中,然后加入8.30g(20mmol)氯化亚锡,氮气保护下回流待溶液颜色变为无色透明时停止反应。用0℃的冰水浴冷却,加入20.0mL的水稀释,用浓氨水调pH至7.5~8.0,用乙酸乙酯萃取3次,无水硫酸钠干燥,减压蒸除溶剂,即得。由于新生成的化合物具有化学活性比较高的基团氨基,其化学结构不稳定易被氧化,因此在用乙酸乙酯萃取后,向其溶液中加入过量的浓盐酸,制备其盐酸盐低温避光备用,待应用时加碱萃取即可使用。
1.4式Ⅰ的芳胺氮芥衍生物的合成
首先合成乙酰氯:在装有回流冷凝管的250mL的三口烧瓶中加入乙酸,在回流冷凝管的一端插入尾气吸收装置,搅拌、升温至70-75℃,再缓慢滴加二氯亚砜,乙酸与二氯亚砜的摩尔比为1:1.5。待反应完成后,减压除去过量的二氯亚砜及反应副产物,即得乙酰氯,对所制备的乙酰氯进行检测,得无色的乙酰氯的纯度大于98%,收率为97.0%,沸点为51℃,密度为1.11g/mL。
在装有回流冷凝管的250mL的三口烧瓶中加入1.00g N,N-二(2-氯乙基)-1,4-苯二胺、20.0mL干燥的二氯甲烷和1.2mL的三乙胺,冰水浴冷却,搅拌下滴加摩尔比为1:5的乙酰氯和二氯甲烷的混合溶液15.0mL,滴加时间约为1.5小时,滴加结束后撤掉冰水浴,常温反应3小时,反应结束后使用30mL的水洗涤反应液,无水硫酸钠干燥,常压蒸馏,滤饼用柱层析(V(石油醚:乙酸乙酯)1∶1)纯化得暗红色晶体,即为最终产物,产物收率为93.0%。
实施例2
1.1 N,N-二(2-羟乙基)-4-硝基苯胺的合成
向250mL三口烧瓶内分别加入摩尔比为1:1.2:0.6的4-氯硝基苯、二乙醇胺和碳酸钾,最后加入质量浓度为1%的CuSO4溶液,且质量浓度为1%的CuSO4溶液与4-氯硝基苯的用量比为0.5mL:3g,混合均匀;加热升高温度,当温度达到82℃时,向反应液中加入甲苯,且甲苯与4-氯硝基苯的用量比为1mL:2g;继续升高温度控制反应温度为120℃,使用薄层色谱法跟踪反应进程,恒温搅拌反应9h。待反应完全后,蒸馏去除甲苯,将蒸馏后的母液倒入60℃的热水中,剧烈搅拌,静置过夜后抽滤除去水层得粗品,粗品用无水乙醇重结晶,得黄色针状晶体N,N-二(2-羟乙基)-4-硝基苯胺。
1.2 N,N-二(2-氯乙基)-4-硝基苯胺的合成
称取2.70g(0.012mol)黄色针状晶体N,N-二(2-羟乙基)-4-硝基苯胺即化合物N,N-二(2-羟基乙基)-4-硝基苯胺,溶于60.0mL干燥的二氯甲烷中,然后向反应瓶中加入1.1mL的三乙胺;取1.3mL(0.018mol)二氯亚砜溶于20.0mL二氯甲烷溶液,室温搅拌下将该溶液滴加到反应瓶,滴加的时间为l.5h,滴加完成后,加热至45℃,回流,并在回流的状态下搅拌反应2h。反应完全后静置冷却,用30.0mL干燥的二氯甲烷稀释,并分别用20.0mL的饱和碳酸氢钠溶液洗涤三次,合并有机层用无水硫酸钠干燥,减压蒸馏除去溶剂,得N,N-二(2-氯乙基)-4-硝基苯胺的粗品。粗品用无水乙醇重结晶得黄色针状晶体N,N-二(2-氯乙基)-4-硝基苯胺2.99g,收率95.0%。
1.3 N,N-二(2-氯乙基)-1,4-苯二胺的合成
称取4mmol黄色针状晶体N,N-二(2-氯乙基)-4-硝基苯胺溶于120.0mL浓盐酸中,然后加入24mmol氯化亚锡,氮气保护下回流待溶液颜色变为无色透明时停止反应。用0℃的冰水浴冷却,加入20.0mL的水稀释,用浓氨水调pH至7.5~8.0,用乙酸乙酯萃取3次,无水硫酸钠干燥,减压蒸除溶剂,即得。由于新生成的化合物具有化学活性比较高的基团氨基,其化学结构不稳定易被氧化,因此在用乙酸乙酯萃取后,向其溶液中加入过量的浓盐酸,制备其盐酸盐低温避光备用,待应用时加碱萃取即可使用。
1.4式Ⅰ的芳胺氮芥衍生物的合成
首先合成乙酰氯:在装有回流冷凝管的250mL的三口烧瓶中加入乙酸,在回流冷凝管的一端插入尾气吸收装置,搅拌、升温至70-75℃,再缓慢滴加二氯亚砜,乙酸与二氯亚砜的摩尔比为1:1.5。待反应完成后,减压除去过量的二氯亚砜及反应副产物,即得乙酰氯,对所制备的乙酰氯进行检测,得无色的乙酰氯的纯度大于98%,收率为97.0%,沸点为51℃,密度为1.11g/mL。
在装有回流冷凝管的250mL的三口烧瓶中加入1.00g N,N-二(2-氯乙基)-1,4-苯二胺、20.0mL干燥的二氯甲烷和1.2mL的三乙胺,冰水浴冷却,搅拌下滴加摩尔比为1:3的乙酰氯和二氯甲烷的混合溶液15.0mL,滴加时间约为2小时,滴加结束后撤掉冰水浴,常温反应8小时,反应结束后使用30mL的水洗涤反应液,无水硫酸钠干燥,常压蒸馏,滤饼用柱层析(V(石油醚:乙酸乙酯)1∶2)纯化得暗红色晶体,即为最终产物,产物收率为93.0%。
实施例3
1.1 N,N-二(2-羟乙基)-4-硝基苯胺的合成
向250mL三口烧瓶内分别加入摩尔比为1:1.4:0.7的4-氯硝基苯、二乙醇胺和碳酸钾,最后加入质量浓度为1%的CuSO4溶液,且质量浓度为1%的CuSO4溶液与4-氯硝基苯的用量比为0.2mL:2g,混合均匀;加热升高温度,当温度达到82℃时,向反应液中加入甲苯,且甲苯与4-氯硝基苯的用量比为1mL:3g;继续升高温度控制反应温度为120℃,使用薄层色谱法跟踪反应进程,恒温搅拌反应7.5h。待反应完全后,蒸馏去除甲苯,将蒸馏后的母液倒入50℃的热水中,剧烈搅拌,静置过夜后抽滤除去水层得粗品,粗品用无水乙醇重结晶,得黄色针状晶体N,N-二(2-羟乙基)-4-硝基苯胺。
1.2 N,N-二(2-氯乙基)-4-硝基苯胺的合成
称取2.70g(0.012mol)黄色针状晶体N,N-二(2-羟乙基)-4-硝基苯胺即化合物N,N-二(2-羟基乙基)-4-硝基苯胺,溶于60.0mL干燥的二氯甲烷中,然后向反应瓶中加入1.1mL的三乙胺;取0.024mol二氯亚砜溶于20.0mL二氯甲烷溶液,室温搅拌下将该溶液滴加到反应瓶,滴加的时间为l.5h,滴加完成后,加热至45℃,回流,并在回流的状态下搅拌反应2h。反应完全后静置冷却,用30.0mL干燥的二氯甲烷稀释,并分别用20.0mL的饱和碳酸氢钠溶液洗涤三次,合并有机层用无水硫酸钠干燥,减压蒸馏除去溶剂,得N,N-二(2-氯乙基)-4-硝基苯胺的粗品。粗品用无水乙醇重结晶得黄色针状晶体N,N-二(2-氯乙基)-4-硝基苯胺2.99g,收率95.0%。
1.3 N,N-二(2-氯乙基)-1,4-苯二胺的合成
称取4mmol黄色针状晶体N,N-二(2-氯乙基)-4-硝基苯胺溶于200.0mL浓盐酸中,然后加入12mmol氯化亚锡,氮气保护下回流待溶液颜色变为无色透明时停止反应。用0℃的冰水浴冷却,加入20.0mL的水稀释,用浓氨水调pH至7.5~8.0,用乙酸乙酯萃取3次,无水硫酸钠干燥,减压蒸除溶剂,即得。由于新生成的化合物具有化学活性比较高的基团氨基,其化学结构不稳定易被氧化,因此在用乙酸乙酯萃取后,向其溶液中加入过量的浓盐酸,制备其盐酸盐低温避光备用,待应用时加碱萃取即可使用。
1.4式Ⅰ的芳胺氮芥衍生物的合成
首先合成乙酰氯:在装有回流冷凝管的250mL的三口烧瓶中加入乙酸,在回流冷凝管的一端插入尾气吸收装置,搅拌、升温至70-75℃,再缓慢滴加二氯亚砜,乙酸与二氯亚砜的摩尔比为1:1.5。待反应完成后,减压除去过量的二氯亚砜及反应副产物,即得乙酰氯,对所制备的乙酰氯进行检测,得无色的乙酰氯的纯度大于98%,收率为97.0%,沸点为51℃,密度为1.11g/mL。
在装有回流冷凝管的250mL的三口烧瓶中加入1.00g N,N-二(2-氯乙基)-1,4-苯二胺、20.0mL干燥的二氯甲烷和1.2mL的三乙胺,冰水浴冷却,搅拌下滴加摩尔比为1:5的乙酰氯和二氯甲烷的混合溶液15.0mL,其中乙酰氯与N,N-二(2-氯乙基)-1,4-苯二胺的摩尔比为1:2;滴加时间约为2小时,滴加结束后撤掉冰水浴,常温反应14小时,反应结束后使用30mL的水洗涤反应液,无水硫酸钠干燥,常压蒸馏,滤饼用柱层析(V(石油醚:乙酸乙酯)1∶3)纯化得暗红色晶体,即为最终产物,产物收率为93.0%。
对上述实施例1制备的N,N-二(2-羟乙基)-4-硝基苯胺进行元素分析,结果如表1所示。
表1 N,N-二(2-羟乙基)-4-硝基苯胺的元素分析结果
对N,N-二(2-羟乙基)-4-硝基苯胺采用溴化钾压片法进行红外分析,结果如图1所示,产物在3282cm-1处出现强吸收峰,属于-OH的伸缩振动所产生的吸收峰;2906cm-1处出现吸收峰,归属亚甲基及中C-H的伸缩振动所产生的吸收峰;1600cm-1、1521cm-1、1483cm-1吸收峰是苯环的骨架振动所产生的吸收峰;1203cm-1处出现的吸收峰属C-N振动吸收峰;819cm-1出现的吸收峰表明为苯环的对二取代C-H弯曲振动引起的吸收峰;1326cm-1处出现的吸收峰为硝基的伸缩振动所产生的吸收峰。综合以上元素分析及红外图谱分析结果确定所合成的化合物为目标化合物N,N-二(2-羟乙基)-4-硝基苯胺。
对上述实施例1制备的N,N-二(2-氯乙基)-4-硝基苯胺进行元素分析,结果如表2所示。
表2元素分析结果
对N,N-二(2-氯乙基)-4-硝基苯胺采用溴化钾压片法进行红外分析,结果如图2所示,对比图1和图2可知,产物在3282cm-1处未出现-OH的伸缩振动吸收峰,且图2较图1在752cm-1及694cm-1处出现了比较强的吸收峰,归属为C-Cl伸缩振动产生的吸收峰;同样在2962cm-1处出现了亚甲基中C-H的伸缩振动吸收峰;1591cm-1、1510cm-1、1494cm-1处的吸收峰是苯环的骨架振动产生的吸收峰;827cm-1处出现的吸收峰表明苯环的对二取代C-H健弯曲振动的吸收峰;1317cm-1处出现的吸收峰为硝基的N-O健伸缩振动产生的吸收峰。综合以上元素分析及红外图谱分析结果确定所合成的化合物为目标化合物N,N-二(2-氯乙基)-4-硝基苯胺。
表3元素分析结果
对上述实施例1制备的N,N-二(2-氯乙基)-1,4-苯二胺进行元素分析,结果如表3所示。
对N,N-二(2-氯乙基)-1,4-苯二胺采用溴化钾压片法进行红外分析,结果如图3所示,对比图3和图2可知,图3在3562cm-1及3442cm-1处出现了两个比较弱的吸收峰,该峰归属伯胺N-H的伸缩振动吸收峰;2962cm-1处强吸收峰,归属亚甲基的C-H伸缩振动吸收峰;1571cm-1、1496cm-1、1458cm-1处的吸收峰归属苯环的骨架振动所产生的吸收峰;819cm-1处的吸收峰表明为苯环的对二取代C-H健弯曲振动的吸收峰;1431cm-1处出现的吸收峰归属C-N伸缩振动吸收峰;图中在2586cm-1及2333cm-1处出现的两个中强吸收峰,归属为胺盐N-H振动吸收峰。综合元素分析结果及红外图谱分析结果确定所合成的化合物为目标化合物N,N-二(2-氯乙基)-1,4苯二胺。
对上述实施例1制备的式Ⅰ的芳胺氮芥衍生物进行元素分析,结果如表4所示。
表4式Ⅰ的芳胺氮芥衍生物的元素分析结果
对上述式Ⅰ的芳氮芥衍生物采用溴化钾压片法进行红外分析,如图4所示,其主要谱带的归属结果如表5所示。
表5式Ⅰ的芳氮芥衍生物的红外光谱主要谱带分析结果
对上述式Ⅰ的芳氮芥衍生物进行核磁分析,用TMS作内标,DMSO作为溶剂,测定酰基化产物的核磁共振氢谱,其核磁图谱见图5,其核磁氢谱数据和结构指派结果见表6。
由图4和表5可知,在3307cm-1处出现的吸收峰归属酰胺基的-NH的伸缩振动吸收峰;1784cm-1~1733cm-1处强的吸收峰属于C=O的伸缩振动吸收峰;2923cm-1处出现强吸收峰,归属甲基及亚甲基中C-H的伸缩振动所产生的吸收峰;1515~1604cm-1又处出现的三个吸收峰是芳环的骨架振动所产生的吸收峰;1456cm-1出现的吸收峰属C-N的伸缩振动吸收峰;721cm-1处出现的吸收峰为C-Cl伸缩振动吸收峰。由此可以初步判断合成的化合物为目标化合物。再由核磁图谱可知,在δH 3.44~3.75之间出现了N,N-二(2-氯乙基)胺中CH2的特征峰,这表明目标产物中含有氮芥基团;δH 6.67~7.41之间有两组苯环上氢的特征吸收峰;δH 1.00出现了-CH3的特征吸收峰;δH 9.69处的特征峰为(NHCO)中氢的特征吸收峰。由红外谱图、核磁谱图及元素分析结果可以确定所合成的化合物为式Ⅰ的芳氮芥衍生物。
表6式Ⅰ的芳氮芥衍生物的核磁氢谱数据分析结果
本发明提出的N,N-二(2-氯乙基)-1,4-苯二胺在低氧环境下有较好的细胞毒性,氨基的供电子作用使氮芥的氮原子上电子云密度增大,可提高氮芥的烷化作用。而肿瘤细胞中存在许多低氧细胞,正常细胞中的细胞相对是富氧的,如果药物只在低氧环境中有细胞毒性,那么这种药物就对肿瘤细胞具有选择性。本发明以N,N-二(2-氯乙基)-1,4-苯二胺为药效基团,以4-氯硝基苯,二乙醇胺为起始原料在碱性物质的存在下,通过亲核取代反应一步合成了N,N-二(2-羟乙基)-4-硝基苯胺,所得化合物与二氯亚砜在三乙胺催化作用下,合成了芳胺氮芥类的主体化合物N,N-二(2-氯乙基)-4-硝基苯胺;以二价锡作为还原剂使N,N-二(2-氯乙基)-4-硝基苯胺4位的硝基还原为氨基得到氮芥类药物的药效体N,N-二(2-氯乙基)-1,4-苯二胺;以乙酰氯对N,N-二(2-氯乙基)-1,4-苯二胺进行结构修饰,使得本发明的芳胺氮芥衍生物具有增强氮芥治疗指数的前提下,又可以有效降低氮芥的毒副作用,同时兼具杀菌、消炎的疗效,以达到降低病人化疗后因免疫力下降而引起的并发症的风险。
虽然,本说明书中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!