一种姜黄醇半抗原及完全抗原的合成方法与流程
本发明属于免疫学领域,具体涉及一种姜黄醇半抗原及完全抗原的合成方法。
背景技术:
姜黄醇又名姜黄环氧醇,目前姜黄醇的测定方法主要有比色法、红外分光光度法、双波长扫描法、气相色谱法等方法,而上述方法在快速、批量检测方面均存在一定的缺陷。免疫分析方法因其具有操作简便、灵敏、稳定、快速和高通量检测的特点,在中药材的质量监测与评价方面具有较好的应用前景。具有与检测成分对应的抗体是免疫分析的关键,姜黄醇为小分子天然化合物,相对分子量236.35,仅具有反应原性,而不具有免疫原性,小分子往往不直接与载体蛋白连接,通过偶联剂与载体蛋白交联。因此需要与大分子蛋白等偶联形成复合物后才能成为完全抗原,但现有技术中完全抗原的产率仅有52%,需要进一步提高产率。
技术实现要素:
针对上述问题,本发明的主要目的在于提供一种姜黄醇半抗原及完全抗原的合成方法,从姜黄醇到最终偶联产物产率得到完全抗原的产率可达90%。
为了达到上述目的,本发明采用如下技术方案:一种姜黄醇半抗原的合成方法,包括如下步骤:
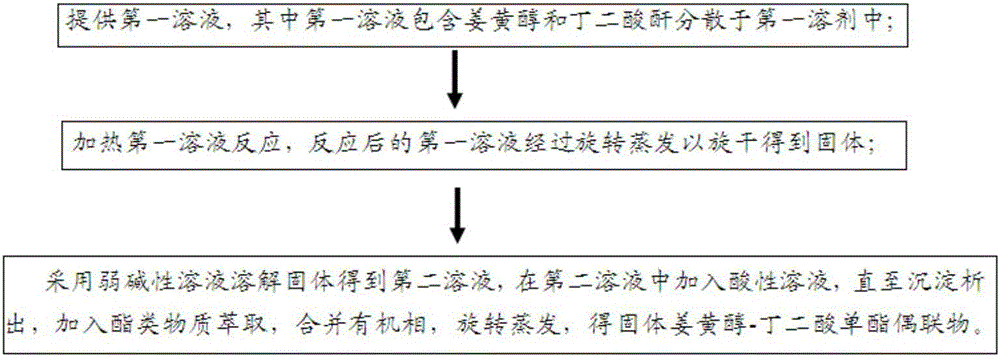
提供第一溶液,其中所述第一溶液包含姜黄醇和丁二酸酐分散于第一溶剂中,所述第一溶剂为吡啶;
加热所述第一溶液反应,反应后的所述第一溶液经旋转蒸发以旋干得到固体;
采用弱碱性溶液溶解所述固体得到第二溶液,在所述第二溶液中加入酸性溶液,直至有沉淀析出,加入酯类物质进行萃取,合并有机相,旋转蒸发以旋干有机相,得固体姜黄醇-丁二酸单酯偶联物。
作为进一步的优选,所述姜黄醇和丁二酸酐在第一溶液中的摩尔比为2:3。
作为进一步的优选,所述姜黄醇和丁二酸酐在第一溶液中的浓度分别为0.05M和0.075M。
作为进一步的优选,所述加热温度为50-80℃。
作为进一步的优选,所述加热温度为70℃。
作为进一步的优选,加热所述第一溶液反应过夜。
作为进一步的优选,所述弱碱性溶液为碳酸氢钠的水溶液,所述碳酸氢钠水溶液的浓度为1%-10%。
作为进一步的优选,所述碳酸氢钠水溶液的浓度为5%。
作为进一步的优选,所述酸性溶液为盐酸溶液。
作为进一步的优选,所述酯类物质选自乙酸乙酯、乙酸异戊酯和苯甲酸甲酯。
作为进一步的优选,所述酯类物质为乙酸乙酯。
一种姜黄醇完全抗原的合成方法,包括如下步骤:
提供第三溶液,其中所述第三溶液包含姜黄醇-丁二酸单酯偶联物分散于第三溶剂中;
提供第四溶液,其中所述第四溶液包含EDC(1-乙基-(3-二甲基氨基丙基)碳酰二亚胺)和NHS(N-羟基琥珀酰亚胺)分散于水中;
将所述第四溶液加入到所述第三溶液中反应得到反应液;
将所述反应液加入到载体蛋白溶液中,反应后透析得到最终偶联产物。
作为进一步的优选,所述第三溶剂为DMF。
作为进一步的优选,所述第三溶液中姜黄醇-丁二酸单酯偶联物的浓度为0.143M。
作为进一步的优选,所述第四溶液中EDC的浓度为0.16-2.6M,所述第四溶液中NHS的浓度为0.087-0.261M。
作为进一步的优选,所述第四溶液中EDC的浓度为0.16M,所述第四溶液中NHS的浓度为0.16M。
作为进一步的优选,所述第四溶液和所述第三溶液的反应温度为4-8℃。
作为进一步的优选,所述第四溶液和所述第三溶液的反应温度为4℃。
作为进一步的优选,所述第四溶液和所述第三溶液反应过夜。
作为进一步的优选,所述载体蛋白溶液的浓度为0.03-0.22mM。
作为进一步的优选,所述载体蛋白溶液的浓度为0.13mM。
作为进一步的优选,所述透析步骤为:透析时间为3d,透析液为1×PBS(磷酸缓冲盐溶液),每天更换透析液三次。
本发明的有益效果是:
(1)本发明采用姜黄醇为原料可制备得到姜黄醇半抗原及完全抗原,根据姜黄醇结构中存在C6羟基的特点,本发明先将姜黄醇制备成姜黄醇琥珀酸单酯(姜黄醇-丁二酸单酯偶联物),然后再与BSA(牛血清白蛋白)或OVA(鸡蛋清白蛋白)偶联形成复合物,从而成为完全抗原,且从姜黄醇到最终偶联产物,得到完全抗原的产率可达90%。
(2)本发明的合成步骤简单,所需的实验条件温和,产物容易得到,且稳定性高。
(3)本发明可制备姜黄醇单克隆抗体,为利用酶联免疫吸附方法对含姜黄醇的中草药进行定性或定量分析提供了可能,可用于大规模生产的中药质量控制、中草药原料药的鉴定、中药活性成分的体内分析及中草药有效成分的纯化等诸多方面,同时为完善中药的质量评价体系和方法起到积极的作用,具有快速、灵敏、精确和简便等特点。
附图说明
图1为本发明实施例姜黄醇半抗原的合成方法的流程示意图。
图2为本发明实施例姜黄醇-HS-BSA的紫外鉴定结果示意图。
图3为本发明实施例姜黄醇-HS-OVA的紫外鉴定结果示意图。
具体实施方式
本发明通过提供一种姜黄醇半抗原及完全抗原的合成方法,解决了现有技术中得到完全抗原产率低的问题。
为了解决上述问题,本发明实施例的主要思路是:
本发明实施例姜黄醇半抗原的合成方法,包括如下步骤:
提供第一溶液,其中所述第一溶液包含姜黄醇和丁二酸酐分散于第一溶剂中,所述第一溶剂为吡啶;
加热所述第一溶液反应,反应后的所述第一溶液经过旋转蒸发以旋干得到固体;
采用弱碱性溶液溶解所述固体得到第二溶液,在所述第二溶液中加入酸性溶液,直至有沉淀析出,加入酯类物质进行萃取,合并有机相,旋转蒸发以旋干有机相,得固体姜黄醇-丁二酸单酯偶联物。
本发明实施例姜黄醇完全抗原的合成方法,包括如下步骤:
提供第三溶液,其中所述第三溶液包含姜黄醇-丁二酸单酯偶联物分散于第三溶剂中;
提供第四溶液,其中所述第四溶液包含EDC和NHS分散于水中;
将所述第四溶液加入到所述第三溶液中反应得到反应液;
将所述反应液加入到载体蛋白溶液中,反应后透析得到最终偶联产物。
本发明实施例所述术语“分散”可理解为溶质溶解在溶剂中,溶解姜黄醇和丁二酸酐的第一溶剂可选为吡啶,吡啶除了作为溶剂,亦有催化姜黄醇和丁二酸酐反应的作用。选择姜黄醇和丁二酸酐物质的用量,使得两者在第一溶剂中呈一定比例的浓度,以达到最佳的反应效果。
本发明实施例所述加热所述第一溶液反应时,一般为加热反应过夜,可采用水浴加热,温度为50-80℃,优选为70℃。反应后的所述第一溶液一般采用旋转蒸发仪以旋干。
所述酸性溶液加入到弱碱性溶液时可采用滴加,并且两者采用适当的浓度,以达到较好的沉淀效果;所述酯类物质为挥发性酯类物质,例如可采用乙酸乙酯、乙酸异戊酯和苯甲酸甲酯等安全且萃取效果好的挥发性物质。
本发明实施例采用的EDC/NHS水溶液可以将反应不稳定的中间产物变稳定,反应活性不变;将所述第四溶液加入到所述第三溶液中反应得到反应液时,其中的加入可采用滴加,控制一定的速度;其中的反应可为反应过夜。
为了让本发明之上述和其它目的、特征、和优点能更明显易懂,下文特举数实施例,来说明本发明所述之姜黄醇半抗原及完全抗原的合成方法。
实施例1
本发明实施例1姜黄醇半抗原衍生物合成步骤如下:
(1)称取姜黄醇118mg(0.5mmol),75mg(0.75mmol)丁二酸酐溶解于10mL吡啶中,70℃水浴加热反应过夜;
(2)取出反应液,用旋转蒸发仪旋干;
(3)用5%NaHCO3的水溶液20mL溶解步骤(2)中已烘干的固体;
(4)用1M HCl滴加入上述步骤(3)溶液中,调pH至2,如有沉淀析出,则继续以下步骤:
(5)加入适量的乙酸乙酯进行萃取,重复3次;
(6)合并有机相,用旋转蒸发仪旋干有机相,得固体,收集后备用;
本发明实施例姜黄醇-HS-BSA合成步骤:
(1)称取姜黄醇-HS(姜黄醇-丁二酸单酯)24mg溶于0.5mL的DMF中,为A液;
(2)取EDC 30mg,NHS 18mg溶于1mL的水中,为B液;
(3)将B液滴入A液中,4℃反应过夜,为C液;
(4)称取BSA 94mg溶于10mL PBS中,将C液加入至BSA溶液中,反应8h后透析3d,每天更换透析液3次(透析液为1×PBS)从姜黄醇到最终偶联产物的产率达到75.6%。
实施例2
姜黄醇半抗原衍生物合成步骤如下:
(1)称取姜黄醇118mg(0.5mmol),75mg(0.75mmol)丁二酸酐溶解于10mL吡啶中,70℃水浴加热反应过夜;
(2)取出反应液,用旋转蒸发仪旋干;
(3)用5%NaHCO3的水溶液20mL溶解已烘干的固体;
(4)用1M HCl滴加入上述溶液中,调pH至2,如有沉淀析出,则继续以下步骤:
(5)加入适量的乙酸乙酯进行萃取,重复3次;
(6)合并有机相,用旋转蒸发仪旋干有机相,得固体,收集后备用;
姜黄醇-HS-OVA合成步骤如下:
(1)称取姜黄醇-HS 24mg溶于0.5mL的DMF中,为A液;
(2)取EDC 30mg,NHS 18mg溶于1mL的水中,为B液;
(3)将B液滴入A液中,4℃反应过夜,为C液;
(4)称取OVA 63mg溶于10mL PBS中,将C液加入至BSA溶液中,反应8h后透析3d,每天更换透析液3次(透析液为1×PBS)从姜黄醇到最终偶联产物的产率达到89.2%。
实施例3
姜黄醇半抗原衍生物合成步骤:
(1)称取姜黄醇118mg(0.5mmol),75mg(0.75mmol)丁二酸酐溶解于10mL吡啶中,80℃水浴加热反应过夜;
(2)取出反应液,用旋转蒸发仪旋干;
(3)用1%NaHCO3 40mL溶解已烘干的固体;
(4)用1M HCl滴加入上述溶液中,调pH至2,如有沉淀析出,则继续以下步骤:
(5)加入适量的乙酸异戊酯进行萃取,重复3次;
(6)合并有机相,用旋转蒸发仪旋干有机相,得固体,收集后备用。
姜黄醇-HS-BSA合成步骤:
(1)称取姜黄醇-HS 24mg溶于0.5mL的DMF中,为A液;
(2)取EDC 300mg,NHS 28mg溶于10mL的水中,为B液;
(3)将B液滴入A液中,4℃反应过夜,为C液;
(4)称取BSA 94mg溶于10mL PBS中,将C液加入至BSA溶液中,反应8h后透析3d,每天更换透析液3次(透析液为1×PBS)从姜黄醇到最终偶联产物的产率达到85.6%。
实施例4
1.姜黄醇半抗原衍生物合成步骤:
(1)称取姜黄醇118mg(0.5mmol),75mg(0.75mmol)丁二酸酐溶解于10mL吡啶中,50℃水浴加热反应过夜;
(2)取出反应液,用旋转蒸发仪旋干;
(3)用10%NaHCO3 15mL溶解已烘干的固体;
(4)用1M HCl滴加入上述溶液中,调pH至2,如有沉淀析出,则继续以下步骤:
(5)加入适量的苯甲酸甲酯进行萃取,重复3次;
(6)合并有机相,用旋转蒸发仪旋干有机相,得固体,收集后备用。
2.姜黄醇-HS-OVA合成步骤:
(1)称取姜黄醇-HS 24mg溶于0.5mL的DMF中,为A液;
(2)取EDC 100mg,NHS 20mg溶于5mL的水中,为B液;
(3)将B液滴入A液中,8℃反应过夜,为C液;
(4)称取OVA63mg溶于10mL PBS中,将C液加入至BSA溶液中,反应8h后透析3d,每天更换透析液3次(透析液为1×PBS)从姜黄醇到最终偶联产物的产率达到90.5%。
实施例5
姜黄醇半抗原衍生物合成步骤:
(1)称取姜黄醇118mg(0.5mmol),75mg(0.75mmol)丁二酸酐溶解于10mL吡啶中,60℃水浴加热反应过夜;
(2)取出反应液,用旋转蒸发仪旋干;
(3)用3%NaHCO3 18mL溶解已烘干的固体;
(4)用1M HCl滴加入上述溶液中,调pH至2,如有沉淀析出,则继续以下步骤:
(5)加入适量的乙酸乙酯进行萃取,重复3次;
(6)合并有机相,用旋转蒸发仪旋干有机相,得固体,收集后备用。
姜黄醇-HS-BSA合成步骤:
(1)称取姜黄醇-HS 24mg溶于0.5mL的DMF中,为A液;
(2)取EDC 200mg,NHS 20mg溶于8mL的水中,为B液;
(3)将B液滴入A液中,6℃反应过夜,为C液;
(4)称取BSA 150mg溶于10mL PBS中,将C液加入至BSA溶液中,反应8h后透析3d,每天更换透析液3次(透析液为1×PBS)从姜黄醇到最终偶联产物的产率达到83.5%。
实施例6
姜黄醇半抗原衍生物合成步骤:
(1)称取姜黄醇118mg(0.5mmol),75mg(0.75mmol)丁二酸酐溶解于10mL吡啶中,70℃水浴加热反应过夜;
(2)取出反应液,用旋转蒸发仪旋干;
(3)用5%NaHCO3 20mL溶解已烘干的固体;
(4)用1M HCl滴加入上述溶液中,调pH至2,如有沉淀析出,则继续以下步骤:
(5)加入适量的乙酸乙酯进行萃取,重复3次;
(6)合并有机相,用旋转蒸发仪旋干有机相,得固体,收集后备用。
姜黄醇-HS-OVA合成步骤:
(1)称取姜黄醇-HS 24mg溶于0.5mL的DMF中,为A液;
(2)取EDC 50mg,NHS 10mg溶于3mL的水中,为B液;
(3)将B液滴入A液中,5℃反应过夜,为C液;
(4)称取OVA 30mg溶于10mLPBS中,将C液加入至BSA溶液中,反应8h后透析3d,每天更换透析液3次(透析液为1×PBS)从姜黄醇到最终偶联产物的产率达到72.6%。
实验例 本实施例1和2中姜黄醇的两种偶联物鉴定:
以紫外分光光度计测定姜黄醇、姜黄醇-HS-BSA和BSA最大吸收峰。如下图2所示:姜黄醇、姜黄醇-HS-BSA和BSA最大吸收波长分别为238nm、274nm和280nm;如下图3所示:姜黄醇、姜黄醇-HS-OVA和OVA最大吸收波长分别为273nm、263nm和275nm。载体偶联本发明实施例半抗原后最大吸收波长发生改变,证明偶联物合成成功。
上述本申请实施例中的技术方案,至少具有如下的技术效果或优点:
(1)本发明采用姜黄醇为原料可制备得到姜黄醇半抗原及完全抗原,根据姜黄醇结构中存在C6羟基的特点,本发明先将姜黄醇制备成姜黄醇琥珀酸单酯,然后再与BSA或OVA偶联形成复合物,从而成为完全抗原,且从姜黄醇到最终偶联产物,得到完全抗原的产率可达90%。
(2)本发明的合成步骤简单,所需的实验条件温和,产物容易得到,且稳定性高。
(3)本发明可制备姜黄醇单克隆抗体,为利用酶联免疫吸附方法对含姜黄醇的中草药进行定性或定量分析提供了可能,可用于大规模生产的中药质量控制、中草药原料药的鉴定、中药活性成分的体内分析及中草药有效成分的纯化等诸多方面,同时为完善中药的质量评价体系和方法起到积极的作用,具有快速、灵敏、精确和简便等特点。
尽管已描述了本发明的优选实施例,但本领域内的技术人员一旦得知了基本创造性概念,则可对这些实施例作出另外的变更和修改。所以,所附权利要求意欲解释为包括优选实施例以及落入本发明范围的所有变更和修改。显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。
- 还没有人留言评论。精彩留言会获得点赞!