高透过率、高折射率含硫光学树脂材料及其制备方法与流程
本发明涉及一种高透光率、高折射含硫光学树脂材料的制备方法,具体涉及制备高纯度1,3,5-三巯甲基苯单体,并与异氰酸酯类化合物进行固化反应,制备高透过率、高折射率光学树脂材料及镜片。
背景技术:
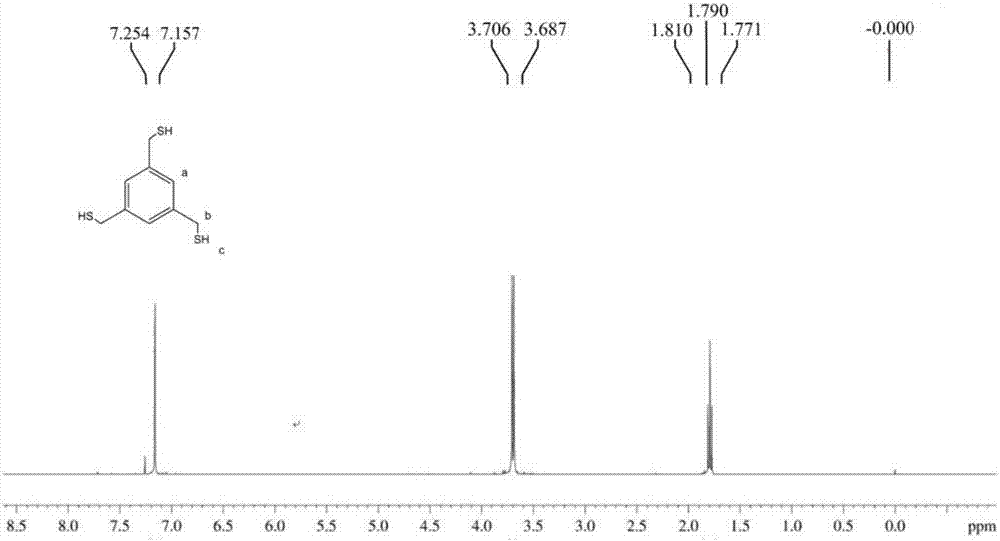
:光学树脂聚合物中含有硫,不仅可以大幅度提高折射率,还能降低色散,增大阿贝数。多元硫醇化合物是一种制备含硫光学树脂的单体,具有高折射、低色散、耐热性耐候性较好等优点。多元硫醇化合物的合成方法有多种,常用的合成方法有硫脲的烃化水解、烯烃与硫化氢加成、硫氢化钠(钾)的烃化、硫醇酯的水解、二硫化物还原、金属有机化合物与硫作用、磺酰氯还原等,例如下列合成方式:1.烯烃与硫化氢反应2.硫脲与溴代烷反应3.二硫化物和碱金属卤化物的制备4.醇与硫化氢反应三元硫醇化合物的折射率、透光率、黄色指数指标受加工温度与合成工艺的影响较大。传统硫脲烃化水解的方法来制备硫醇时,由于有些物质在反应过程中生成了一取代硫醇、二取代硫醇和偶氮结构类副产物,产物泛黄,影响硫醇单体的折射率、透过率和纯度,作为光学材料,不适合制造高精密透镜和光学器件。技术实现要素:本发明通过制备高纯度1,3,5-三巯甲基苯单体,并与异氰酸酯类化合物进行固化反应,制备高透过率、高折射率光学树脂材料及镜片。本发明所述方法制备产品折射率、透光率高、黄色指数低。为实现上述目的,本发明提供如下技术方案:一种高透过率、高折射率含硫光学树脂材料的制备方法,该方法包含以下步骤:i.制备异硫脲盐:向反应容器中加入1,3,5-三溴甲基苯和乙醇-乙酸正丁脂混合溶剂,室温下搅拌分散均匀;其中乙醇、乙酸正丁脂按(5.5-6.5)∶1质量比混合,1,3,5-三溴甲基苯与混合溶剂的质量比为1∶(20-30);随后加入硫脲,搅拌20-24小时,硫脲与1,3,5-三溴甲基苯质量比为(0.5-1.2)∶1,溶液中析出大量白色沉淀;过滤,沉淀用乙醇-乙酸正丁脂混合溶液洗涤过滤2-3次,该乙醇-乙酸正丁脂混合溶液中乙醇和乙酸正丁脂按(4.5-5.5)∶1质量比例混合,干燥后,得到白色异硫脲盐;ii.加碱水解:异硫脲盐加入20-30倍重量的蒸馏水中,搅拌溶解至澄清,再加入氢氧化钠片碱,氢氧化钠与步骤i所用的1,3,5-三溴甲基苯的质量比为(0.5-1)∶1,加热回流4~5小时,冷却至室温;iii.酸化生成硫醇:用冰浴使体系降温至10~20℃,滴加盐酸调节体系ph值至3-3.5,用三氯甲烷萃取,得到含三元硫醇的有机相溶液,将有机相溶液用蒸馏水洗至中性;iv.脱色纯化:步骤iii获得的溶液干燥除水后,向其中加入凹凸棒土吸附剂,吸附剂的加入量与步骤i所用硫脲等质量,搅拌混合35-45分钟后,静置2-3小时,溶液中少量偶氮类生色团被吸附,去除凹凸棒土吸附剂,蒸除溶剂后,得到高纯度无色1,3,5-三巯甲基苯;v.固化反应:以二月桂酸二丁基锡为催化剂,将步骤iv获得的1,3,5-三巯甲基苯与异氰酸酯进行固化反应制备高透过率、高折射率含硫光学树脂材料。如上所述的制备方法,其特征在于,所述凹凸棒土吸附剂的粒径为100~400目。如上所述的制备方法,其特征在于,所述步骤v的具体操作为:将1,3,5-三巯甲基苯和异氰酸酯类化合物按照摩尔比为(0.7~1.3)∶(1~2)混合;加入相对于1,3,5-三巯甲基苯1-3%重量的催化剂二月桂酸二丁基锡,加入助剂,然后将上述混合物于58-62℃下预聚合35-45min,真空脱泡后将原料浇注到模具中,将原料升到90~130℃使其固化成型,随后缓慢降温到室温,脱模清洗后得到无色透明光学树脂材料。如上所述的制备方法,其特征在于,所述1,3,5-三巯甲基苯和异氰酸酯类化合物的摩尔比为(0.7~1)∶(1~1.5)。如上所述的制备方法,其特征在于,所述异氰酸酯类化合物为甲苯二异氰酸酯、二苯基甲烷-4,4’-二异氰酸酯、1.6-己二异氰酸酯、间苯二亚甲基二异氰酸酯、萘-1.5-二异氰酸酯、甲基环己基二异氰酸酯、二环己基甲烷二异氰酸酯、四甲基苯二亚甲基二异氰酸酯和异佛尔酮二异氰酸酯中的一种或一种以上混合。如上所述的制备方法,其特征在于,所述方法包含以下操作步骤:i.制备异硫脲盐:向反应容器中加入1,3,5-三溴甲基苯和乙醇-乙酸正丁脂混合溶剂,室温下搅拌分散均匀;其中乙醇、乙酸正丁脂按6∶1质量比混合,1,3,5-三溴甲基苯与混合溶剂的质量比为1∶25;随后加入硫脲,搅拌24小时,硫脲与1,3,5-三溴甲基苯质量比为3∶4,溶液中析出大量白色沉淀;过滤,沉淀用乙醇-乙酸正丁脂混合溶液洗涤过滤3次,该乙醇-乙酸正丁脂混合溶液中乙醇和乙酸正丁脂按5∶1质量比例混合,干燥后,得到白色异硫脲盐;ii.加碱水解:异硫脲盐加入25倍重量的蒸馏水中,搅拌溶解至澄清,再加入氢氧化钠片碱,氢氧化钠与步骤i所用的1,3,5-三溴甲基苯的质量比为3∶4,加热回流5小时,冷却至室温;iii.酸化生成硫醇:用冰浴使体系降温至20℃以下,滴加盐酸调节体系ph值至3,用三氯甲烷萃取,得到含三元硫醇的有机相溶液,将有机相溶液用蒸馏水洗至中性;iv.脱色纯化:步骤iii获得的溶液干燥除水后,向其中加入凹凸棒土吸附剂,吸附剂的加入量与步骤i所用硫脲等质量,搅拌混合40分钟后,静置2.5小时,溶液中少量偶氮类生色团被吸附,去除凹凸棒土吸附剂,蒸除溶剂后,得到高纯度无色1,3,5-三巯甲基苯;v.固化反应:将1,3,5-三巯甲基苯和异氰酸酯类化合物按照摩尔比为(0.7~1)∶(1~1.5)混合;加入相对于1,3,5-三巯甲基苯1-3%重量的催化剂二月桂酸二丁基锡,加入助剂,然后将上述混合物于60℃下预聚合40min,真空脱泡后将原料浇注到模具中,将原料升到90~130℃使其固化成型,随后缓慢降温到室温,脱模清洗后得到无色透明光学树脂材料。另一方面,本发明提供一种高透过率、高折射率含硫光学树脂材料,其是采用如上所述的方法制备的。又一方面,本发明提供一种高透过率、高折射率含硫光学树脂镜片,其是采用如上所述的方法制备的。本发明中1,3,5-三巯甲基苯单体的合成路线如下:本发明1,3,5-三巯甲基苯的合成方法中,制备异硫脲盐步骤采用乙醇-乙酸正丁脂混合溶剂。由于三取代异硫脲盐不易溶于乙醇-乙酸正丁脂混合溶剂,但是一取代硫脲盐、二取代硫脲盐、溴甲基可以完全溶解,由此可以有效分离一取代硫脲盐、二取代硫脲盐副产物,但得到具有极高纯度的产物。实验过程中可以观察到当三溴甲基苯和硫脲反应后,体系澄清,继续反应一段时间生成三取代异硫脲盐后,就会析出大量纯白色固体。另外,在加热过程中产生了极少偶氮类生色团,使产物泛黄,影响产品纯度和透明度,经反复试验,选用凹凸棒土吸附剂去除副产物,效果明显。本发明的有益效果在于,本发明方法制备的1,3,5-三巯甲基苯纯度高,黄色指数低,由其制备的异氰酸酯树脂镜片透光率高、折射率高,黄色指数低。其中1,3,5-三巯甲基苯透光率≥89%,折射率≥1.65,黄色指数≤1;树脂镜片透光率≥90%,折射率≥1.66,黄色指数≤1.50。附图说明图1为实施例1制备的1,3,5-三巯甲基苯的ft--ir红外光谱图。图2为实施例1制备的1,3,5-三巯甲基苯的hnmr核磁共振氢谱图。图3为实施例1制备的1,3,5-三巯甲基苯的cnmr核磁共振碳谱图。图4为1,3,5-三巯甲基苯透过率光谱对比图。图5为树脂镜片透过率光谱对比图。具体实施方式下面通过具体实施例对本发明做进一步说明,但并不意味着对本发明保护范围的限制。实施例1:制备1,3,5-三巯甲基苯三元硫醇单体与光学树脂镜片(1)制备1,3,5-三巯甲基苯:在四口瓶中加入1,3,5-三溴甲基苯40g,加入乙醇和乙酸正丁脂混合溶液1000ml,其中乙醇和乙酸正丁脂按质量比6∶1配成,磁子搅拌分散均匀后,再加入硫脲30g,搅拌24小时,待析出大量白色沉淀,过滤用乙醇和乙酸正丁脂按质量比5∶1混合,洗涤过滤3次,干燥后,得到白色异硫脲盐;继续加入1000ml蒸馏水,搅拌溶解至澄清,再加入naoh30g,加热回流5小时,冷却至室温;用冰浴使体系降温后,滴加盐酸,调节体系ph值至3,用三氯甲烷300ml×3萃取,得到含三元硫醇的有机相溶液,将有机相溶液用蒸馏水洗至中性;脱水后搅拌加入30g凹凸棒土吸附剂,搅拌混合40分钟后,静止2.5小时,溶液中少量偶氮类生色团被吸附,分层去除下层凹凸棒土吸附剂,蒸除溶剂后,得到15.6g高纯度无色1,3,5-三巯甲基苯。红外光谱检测分析:1,3,5-三巯甲基苯在溴化钾盐片上涂膜后测得红外光谱(图1),2554.26cm-1处的吸收峰是液态脂肪族硫醇的s-h键的伸缩振动峰,这是鉴定s-h基团的特征峰,验证了这一步反应生成了巯基;708.37cm-1处的吸收峰则是c-s键的伸缩振动;1600.67cm-1和1453.50cm-1两处的吸收峰以双峰形式出现,是苯环的骨架振动,包括环内碳碳伸缩振动等;3000cm-1附近的吸收峰则是苯环上的c-h伸缩振动。核磁共振氢谱检测分析:将1,3,5-三巯甲基苯溶于氘代氯仿后,测试得到核磁共振氢谱(图2),δ=1.77~1.81ppm,δ=3.69~3.71ppm,δ=7.16ppm,三种化学位移的氢的个数比约为1∶2∶1,根据化学位移的大小及氢的个数比推断:δ=7.16ppm对应于三元硫醇分子中a处苯环上相连的一个氢,因相邻碳上没有氢原子而呈单峰;δ=3.69~3.71ppm对应b处亚甲基上的两个氢,受到相邻硫原子连有一个氢的影响而呈双峰;δ=1.77~1.81ppm则对应于c处巯基上的一个氢,受相邻亚甲基上两个氢的影响而呈三重峰。核磁共振碳谱检测分析:将1,3,5-三巯甲基苯溶于氘代氯仿后,测试得到核磁共振碳谱(图3),δ=76.89~77.53ppm为氘代氯仿的溶剂峰。δ=126.56ppm对应三元硫醇分子中a处苯环上与氢相连的碳原子;δ=142.04ppm对应于b处苯环上与亚甲基相连的碳原子,受巯甲基影响而使化学位移偏向于低场;δ=28.75ppm处于高场,则对应于c处亚甲基上的碳原子。通过以上综合分析证实已合成出了目的产物1,3,5-三巯甲基苯。(2)制备高折射率光学树脂镜片:将1,3,5-三巯甲基苯三元硫醇200g与280g异氰酸酯化合物混合搅拌,加入3g二月桂酸二丁基锡(dbtl)催化剂,然后将上述混合物于60℃下预聚合40min,真空脱泡后将原料浇注到模具中,按照固化程序将原料升到120℃使得其固化成型,随后缓慢降温到室温,脱模清洗后得到无色透明光学镜片。实施例2:制备1,3,5-三巯甲基苯与光学树脂镜片(1)制备1,3,5-三巯甲基苯:在四口瓶中加入1,3,5-三溴甲基苯40g,加入乙醇和乙酸正丁脂混合溶液800ml,其中乙醇和乙酸正丁脂按质量比6∶1配成,磁子搅拌分散均匀后,再加入硫脲20g,搅拌24小时,待析出大量白色沉淀,过滤用乙醇和乙酸正丁脂按质量比5∶1混合,洗涤过滤2-3次,干燥后,得到白色异硫脲盐;继续加入800ml蒸馏水,搅拌溶解至澄清,再加入naoh20g,加热回流4小时,冷却至室温;用冰浴使体系降温后,滴加盐酸,调节体系ph值至3,用三氯甲烷260ml×3萃取,得到含三元硫醇的有机相溶液,将有机相溶液用蒸馏水洗至中性;脱水后向三元硫醇里搅拌加入20g凹凸棒土吸附剂,搅拌混合40分钟后,静止2.5小时,溶液中少量偶氮类生色团被吸附,分层去除下层凹凸棒土吸附剂,蒸除溶剂后,得到15.2g高纯度无色1,3,5-三巯甲基苯。红外光谱、核磁共振氢谱的测试结果与实施例1相似。(2)制备高折射率光学树脂镜片:将1,3,5-三巯甲基苯三元硫醇200g与400g异氰酸酯化合物混合搅拌,加入4g二月桂酸二丁基锡(dbtl)催化剂,然后将上述混合物于60℃下预聚合40min,真空脱泡后将原料浇注到模具中,按照固化程序将原料升到110℃使得其固化成型,随后缓慢降温到室温,脱模清洗后得到无色透明光学镜片。实施例3:制备1,3,5-三巯甲基苯与光学树脂镜片(1)制备1,3,5-三巯甲基苯:在四口瓶中加入1,3,5-三溴甲基苯40g,加入乙醇和乙酸正丁脂混合溶液1200ml,其中乙醇和乙酸正丁脂按质量比6∶1配成,磁子搅拌分散均匀后,再加入硫脲50g,搅拌24小时,待析出大量白色沉淀,过滤用乙醇和乙酸正丁脂按质量比5∶1混合,洗涤过滤2-3次,干燥后,得到白色异硫脲盐;继续加入1200ml蒸馏水,搅拌溶解至澄清,再加入naoh40g,加热回流5小时,冷却至室温;用冰浴使体系降温后,滴加盐酸,调节体系ph值至3,用三氯甲烷400ml×3萃取,得到含三元硫醇的有机相溶液,将有机相溶液用蒸馏水洗至中性;脱水后搅拌加入50g凹凸棒土吸附剂,搅拌混合40分钟后,静止2.5小时,溶液中少量偶氮类生色团被吸附,分层去除下层凹凸棒土吸附剂,蒸除溶剂后,得到15.8g高纯度无色1,3,5-三巯甲基苯。红外光谱、核磁共振氢谱的测试结果与实施例1相似。(2)制备高折射率光学树脂镜片:将1,3,5-三巯甲基苯三元硫醇200g与230g异氰酸酯化合物混合搅拌,加入5g二月桂酸二丁基锡(dbtl)催化剂,然后将上述混合物于60℃下预聚合40min,真空脱泡后将原料浇注到模具中,按照固化程序将原料升到100℃,使得其固化成型,随后缓慢降温到室温,脱模清洗后得到无色透明光学镜片。实施例4:制备1,3,5-三巯甲基苯与光学树脂镜片(1)制备1,3,5-三巯甲基苯:在四口瓶中加入1,3,5-三溴甲基苯40g,加入乙醇和乙酸正丁脂混合溶液900ml,其中乙醇和乙酸正丁脂按质量比6∶1配成,磁子搅拌分散均匀后,再加入硫脲26g,搅拌24小时,待析出大量白色沉淀,过滤用乙醇和乙酸正丁脂按质量比5∶1混合,洗涤过滤2-3次,干燥后,得到白色异硫脲盐;继续加入900ml蒸馏水,搅拌溶解至澄清,再加入naoh25g,加热回流4小时,冷却至室温;用冰浴使体系降温后,滴加盐酸,调节体系ph值至3,用三氯甲烷280ml×3萃取,得到含三元硫醇的有机相溶液,将有机相溶液用蒸馏水洗至中性;脱水后向三元硫醇里加入搅拌加入26g凹凸棒土吸附剂,搅拌混合40分钟后,静止2.5小时,溶液中少量偶氮类生色团被吸附,分层去除下层凹凸棒土吸附剂,蒸除溶剂后,得到15.5高纯度无色1,3,5-三巯甲基苯。红外光谱、核磁共振氢谱的测试结果与实施例1相似。(2)制备高折射率光学树脂镜片:将1,3,5-三巯甲基苯三元硫醇200g与320g异氰酸酯化合物混合搅拌,加入2g二月桂酸二丁基锡(dbtl)催化剂,然后将上述混合物于60℃下预聚合40min,真空脱泡后将原料浇注到模具中,按照固化程序将原料升到95℃,使其固化成型,随后缓慢降温到室温,脱模清洗后得到无色透明光学镜片。实施例5:制备1,3,5-三巯甲基苯与光学树脂镜片(1)1,3,5-三巯甲基苯:在四口瓶中加入1,3,5-三溴甲基苯40g,加入乙醇和乙酸正丁脂混合溶液1100ml,其中乙醇和乙酸正丁脂按质量比6∶1配成,磁子搅拌分散均匀后,再加入硫脲40g,搅拌24小时,待析出大量白色沉淀,过滤用乙醇和乙酸正丁脂按质量比5∶1混合,洗涤过滤2-3次,干燥后,得到白色异硫脲盐;继续加入1100ml蒸馏水,搅拌溶解至澄清,再加入naoh35g,加热回流5小时,冷却至室温;用冰浴使体系降温后,滴加盐酸,调节体系ph值至3,用三氯甲烷350ml×3萃取,得到含三元硫醇的有机相溶液,将有机相溶液用蒸馏水洗至中性;脱水后向三元硫醇里搅拌加入40g凹凸棒土吸附剂,搅拌混合40分钟后,静止2.5小时,溶液中少量偶氮类生色团被吸附,分层去除下层凹凸棒土吸附剂,蒸除溶剂后,得到15.7g高纯度无色1,3,5-三巯甲基苯。红外光谱、核磁共振氢谱的测试结果与实施例1相似。(2)制备高折射率光学树脂镜片:将1,3,5-三巯甲基苯三元硫醇200g与200g异氰酸酯化合物混合搅拌,加入5.5g二月桂酸二丁基锡(dbtl)催化剂,然后将上述混合物于60℃下预聚合40min,真空脱泡后将原料浇注到模具中,按照固化程序将原料升到120℃,使其固化成型,随后缓慢降温到室温,脱模清洗后得到无色透明光学镜片。对比例1:采用甲醇溶剂制备1,3,5-三巯甲基苯与光学树脂镜片(1)制备1,3,5-三巯甲基苯:在四口瓶中加入1,3,5-三溴甲基苯40g,加入甲醇1000ml,磁子搅拌分散均匀后,再加入硫脲30g,搅拌24小时,待析出大量白色沉淀,过滤用甲醇洗涤过滤2-3次,干燥后,得到白色异硫脲盐;继续加入1000ml蒸馏水,搅拌溶解至澄清,再加入naoh30g,加热回流4~5小时,冷却至室温;用冰浴使体系降温后,滴加盐酸,调节体系ph值至3,用三氯甲烷300ml×3萃取,得到含三元硫醇的有机相溶液,将有机相溶液用蒸馏水洗至中性,用无水硫酸钠干燥,过滤后旋蒸得到19.2克1,3,5-三巯甲基苯。(2)制备高折射率光学树脂镜片:将1,3,5-三巯甲基苯三元硫醇200g与280g异氰酸酯化合物混合搅拌,加入3g二月桂酸二丁基锡(dbtl)催化剂,然后将上述混合物于60℃下预聚合40min,真空脱泡后将原料浇注到模具中,按照固化程序将原料升到120℃,使其固化成型,随后缓慢降温到室温,脱模清洗后得到无色透明光学镜片。对比例2:采用乙醇溶剂制备1,3,5-三巯甲基苯与光学树脂镜片(1)制备1,3,5-三巯甲基苯:在四口瓶中加入1,3,5-三溴甲基苯40g,加入1000ml乙醇,磁子搅拌分散均匀后,再加入硫脲30g,搅拌24小时,待析出大量白色沉淀,过滤用乙醇洗涤过滤2-3次,干燥后,得到白色异硫脲盐;继续加入1000ml蒸馏水,搅拌溶解至澄清,再加入naoh30g,加热回流4~5小时,冷却至室温;用冰浴使体系降温后,滴加盐酸,调节体系ph值至3,用三氯甲烷300ml×3萃取,得到含三元硫醇的有机相溶液,将有机相溶液用蒸馏水洗至中性,用无水硫酸钠干燥,过滤后旋蒸得到17.9g1,3,5-三巯甲基苯。(2)制备高折射率光学树脂镜片:将1,3,5-三巯甲基苯三元硫醇200g与280g异氰酸酯化合物混合搅拌,加入4g二月桂酸二丁基锡(dbtl)催化剂,然后将上述混合物于60℃下预聚合40min,真空脱泡后将原料浇注到模具中,按照固化程序将原料升到115℃,使其固化成型,随后缓慢降温到室温,脱模清洗后得到无色透明光学镜片。实施例6对实施例1-5和对比例1-2制备的分别进行光学性能检测,其中透过率检测选用上海元析仪器有限公司uv-8000型紫外可见光光度计,检测方法:直接将树脂单体涂在紫外可见光光度计的棱镜上测定透光率;折射率和阿贝数检测选用上海光学仪器设备有限公司wzs1型阿贝折射仪,检测方法:直接将树脂单体涂在阿贝折射仪的棱镜上测定折射率和阿贝数;黄色指数由分光光度计的读数根据公式计算而得。公式:式中,yi为黄色指数;x、y、z是c光源的三刺激值;表面硬度检测:树脂样品的表面硬度用铅笔硬度表示,参考国家标准,采用中华高级绘图铅笔,笔芯直径1mm,笔尖磨平,测试时以1kg的力沿45℃向前推,以无划痕的最大硬度的铅笔硬度为样品的表面硬度。检测结果列于下表(表一、表二)。从检测结果得知:本发明制造的1,3,5-三巯甲基苯透光率≥89%,折射率≥1.65,黄色指数≤1;镜片透光率≥90%,折射率≥1.66,黄色指数≤1.50。表一实施例与对比例三元硫醇单体光学性能检测结果表二实施例与对比例光学镜片性能检测结果样品透过率(t%)折射率(nd)黄色指数(yi)表面硬度(h)实施例190.21.6641.273h实施例290.21.6601.293h实施例390.11.6631.473h实施例490.31.6621.353h实施例590.21.6641.423h对比例188.61.6412.193h对比例289.41.6592.063h当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1