一种合成多取代嘧啶杂环化合物的方法与流程
本发明涉及医药合成技术领域,具体涉及到一种合成多取代嘧啶杂环化合物的方法。
背景技术:
嘧啶类杂环化合物是是一类具有生物活性和药物活性的化合物,也是众多活性分子比如药物,生物碱的合成中间体,广泛应用于有机合成、药物、天然产物、材料等领域;尤其是在农业和医药方面具有广泛应用。嘧啶类杂环化合物在农业生产中发挥极大的作用(徐英,杨吉春,任兰会,等.具有杀虫活性的嘧啶类化合物的研究进展[A].农药,2011,7(50):474-478.),被广泛用作杀虫剂、除草剂和杀菌剂等(如抗蚜威、环虫腈等);其在医药上也得到了广泛的应用,例如5-氟尿嘧啶是临床广泛使用的抗代谢、抗肿瘤药物;一些1-(2-羟乙氧基)甲基-6-(苯硫基)胸腺嘧啶类化合物能够较强的抑制HIV病毒,具有广泛的抗耐药作用。
近年来,利用过渡金属和纳米催化构建嘧啶杂环的强有力的手段,这些方法也相继被报道,其中主要有(1)铜催化(N.Chernyak,V.Gevorgyan,Angew.Chem.,Int.Ed.2010,49,2743;P.Liu,L.-S.Fang,X.Lei,G.-Q.Lin,Tetrahedron Lett.2010,51,4605;S.Mishra,R.Ghosh,Synthesis 2011,3463;S.K.Guchhait,A.L.Chandgude,G.Priyadarshani,J.Org.Chem.2012,77,4438);(2)铟催化(B.V.S.Reddy,P.S.Reddy,Y.J.Reddy,J.S.Yadav,Tetrahedron Lett.2011,52,5789);(3)纳米催化(Ali Maleki*(*表示通讯作者),Helvetica Chimica Acta.2014,97,587)。而无金属催化多组分反应生成嘧啶类杂环化合物的方法目前还没有报道。
技术实现要素:
本发明提供了一种多取代嘧啶杂环化合物的新方法,该方法操作简单便捷,反应过程不需要过渡金属催化剂,商品化的原料廉价易得无污染,污染小,环境友好。
本发明的原理是以脒、醛,炔作为原料,在碱的作用下,产生炔醇中间体,然后脱水缩合,一步合成了多取代嘧啶杂环化合物。且原料廉价易得,方法简单易行,操作安全,因而具有很高的潜在实用价值。
本发明的目的通过以下技术方案实现:
一种合成多取代嘧啶杂环化合物的方法:在反应容器中,以脒,炔,醛为原料,以碱为促进剂,以有机溶剂为溶剂,反应,冷却,萃取,去除溶剂,提纯,得到嘧啶杂环化合物。
上述反应如下式所示:
所述脒的结构为所述炔的结构为:所述醛的结构为:R3CHO;
所述嘧啶杂环化合物的结构式为
其中,R1为取代或未取代的芳香基团、饱和或不饱和含有1~20个碳原子的线性或支链烃基基团、氨基,优选为苯基,对溴苯基,间溴苯基,对氯苯基,邻氯苯基,对氟苯基,对甲苯基,间甲苯基,邻甲苯基,间甲氧基苯基,对甲氧基苯基,氨基,甲基,环丙基,甲氨基或4-吡啶基中一种。
R2为取代或未取代的芳香基团、饱和或不饱和含有1~20个碳原子的线性或支链烃基基团,优选为苯基、对戊基苯基,对溴苯基,对甲苯基,对甲氧基苯基,邻氯苯基,间甲氧基苯基,间三氟甲基苯基,邻氟苯基,对氯苯基,间氯苯基或邻甲氧基苯基。
R3为取代或未取代的芳香基团、饱和或不饱和含有1~20个碳原子的线性或支链烃基基团,优选为对甲氧基苯基,邻甲氧基苯基,对甲苯基,邻氯苯基,苯基,间氟苯基,对氯苯基,4-吡啶基,2-呋喃基,2,4-二甲基苯基或对叔丁基苯基。
上述方法中:脒与炔、醛的摩尔比为1:(1~3):(1~3);加入的碱量与脒的摩尔比为(1~3):1。
上述方法中:所述的碱为有机碱或无机碱,优选为叔丁醇锂、碳酸铯、碳酸钾、碳酸钠、甲醇钠、三乙胺、氢氧化钾、磷酸钾、碳酸氢钠、叔丁醇钾、氢氧化钠中一种以上。
上述方法中:所述的溶剂为有机溶剂,优选为甲苯、二甲苯、乙腈、1,2-二氯乙烷、四氢呋喃、N,N-二甲基甲酰胺、二甲基亚砜、1,4-二氧六环、乙醇、N,N-二甲基乙酰胺、硝基甲烷中一种以上。
所述反应的条件为在50~160℃下搅拌3~24小时,所述冷却的温度为冷却至室温,所述去除溶剂为减压蒸除溶剂,所述提纯是指经柱层析提纯。
所述柱层析的洗脱液为石油醚和乙酸乙酯的混合溶剂;石油醚和乙酸乙酯的体积比为(5~500):1。
所述嘧啶杂环化合物通过上述方法制备得到。
本发明相对于现有的技术,具有以下优点及效果:
本发明一种合成多取代嘧啶杂环化合物的新合成方法具有以下优点,操作安全简单,所用原料无毒、廉价易得,环境友好,对底物适应性广、产率优良,且反应条件温和,无金属催化,因而具有良好的工业应用前景。
附图说明
图1和图2分别是实施例7所得产物氢谱图和碳谱图;
图3和图4分别是实施例8所得产物氢谱图和碳谱图;
图5和图6分别是实施例9所得产物氢谱图和碳谱图;
图7和图8分别是实施例10所得产物氢谱图和碳谱图;
图9和图10分别是实施例11所得产物氢谱图和碳谱图;
图11和图12分别是实施例12所得产物氢谱图和碳谱图;
图13和图14分别是实施例13所得产物氢谱图和碳谱图;
图15和图16分别是实施例14所得产物氢谱图和碳谱图;
图17和图18分别是实施例15所得产物氢谱图和碳谱图;
图19和图20分别是实施例16所得产物氢谱图和碳谱图;
图21和图22分别是实施例17所得产物氢谱图和碳谱图;
图23和图24分别是实施例18所得产物氢谱图和碳谱图;
图25和图26分别是实施例19所得产物氢谱图和碳谱图;
图27和图28分别是实施例20所得产物氢谱图和碳谱图;
图29和图30分别是实施例21所得产物氢谱图和碳谱图。
具体实施方式
下面结合具体实施例和附图,对本发明作进一步的详细说明,但本发明的实施方式和适应的底物不限于此。
实施例1
在螺口试管中加入0.20毫摩尔苯甲脒盐酸盐、0.25毫摩尔苯乙炔、0.25毫摩尔2,4-二甲基苯甲醛、0.4毫摩尔三乙胺、1毫升二甲基亚砜,空气条件下,在120℃搅拌反应12小时,停止加热及搅拌,冷却至室温。乙酸乙酯萃取反应液,减压旋蒸去除溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为100:1的石油醚:乙酸乙酯混合溶剂,产率为24%。
实施例2
在螺口试管中加入0.20毫摩尔苯甲脒盐酸盐、0.25毫摩尔苯乙炔、0.25毫摩尔2,4-二甲基苯甲醛、0.4毫摩尔氢氧化钾、1毫升二甲基亚砜,空气条件下,在120℃搅拌反应12小时,停止加热及搅拌,冷却至室温。乙酸乙酯萃取反应液,减压旋蒸去除溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为100:1的石油醚:乙酸乙酯混合溶剂,产率为55%。
实施例3
在螺口试管中加入0.20毫摩尔苯甲脒盐酸盐、0.25毫摩尔苯乙炔、0.25毫摩尔2,4-二甲基苯甲醛、0.4毫摩尔叔丁醇钾,1毫升乙腈,空气条件下,在80℃搅拌反应12小时,停止加热及搅拌,冷却至室温。乙酸乙酯萃取反应液,减压旋蒸去除溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为100:1的石油醚:乙酸乙酯混合溶剂,产率为47%。
实施例4
在螺口试管中加入0.20毫摩尔苯甲脒盐酸盐、0.25毫摩尔苯乙炔、0.25毫摩尔2,4-二甲基苯甲醛、0.4毫摩尔叔丁醇钾,1毫升乙醇,空气条件下,在80℃搅拌反应12小时,停止加热及搅拌,冷却至室温。乙酸乙酯萃取反应液,减压旋蒸去除溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为100:1的石油醚:乙酸乙酯混合溶剂,产率为44%。
实施例5
在螺口试管中加入0.20毫摩尔苯甲脒盐酸盐、0.25毫摩尔苯乙炔、0.25毫摩尔2,4-二甲基苯甲醛、0.4毫摩尔叔丁醇钾,1毫升二甲基亚砜,真空条件下,在120℃搅拌反应12小时,停止加热及搅拌,冷却至室温。乙酸乙酯萃取反应液,减压旋蒸去除溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为100:1的石油醚:乙酸乙酯混合溶剂,产率为55%。
实施例6
在螺口试管中加入0.20毫摩尔苯甲脒盐酸盐、0.25毫摩尔苯乙炔、0.25毫摩尔2,4-二甲基苯甲醛、0.4毫摩尔叔丁醇钾,1毫升二甲基亚砜,氧气条件下,在120℃搅拌反应12小时,停止加热及搅拌,冷却至室温。乙酸乙酯萃取反应液,减压旋蒸去除溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为100:1的石油醚:乙酸乙酯混合溶剂,产率为50%。
实施例7
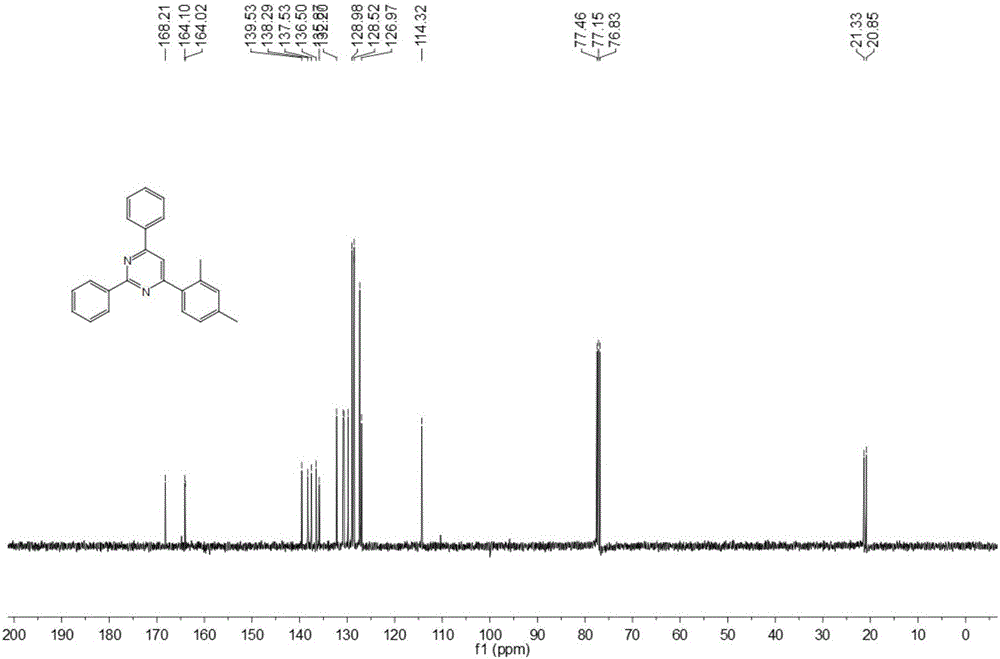
在螺口试管中加入0.20毫摩尔苯甲脒盐酸盐、0.25毫摩尔苯乙炔、0.25毫摩尔2,4-二甲基苯甲醛、0.4毫摩尔叔丁醇钾,1毫升二甲基亚砜,氮气保护条件下,在120℃搅拌反应12小时,停止加热及搅拌,冷却至室温。乙酸乙酯萃取反应液,减压旋蒸去除溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为100:1的石油醚:乙酸乙酯混合溶剂,产率为73%。
本实施例所得化合物的氢谱图和碳谱图分别如图1和图2所示,其结构表征数据如下:
IR(KBr)2922,2855,1735,1630,1520,1455,1384,1120,879,772,598cm-1;
1H NMR(400MHz,CDCl3,ppm):δ8.67(d,J=8.0MHz,2H),8.24(d,J=8.0MHz,2H),7.68(s,1H),7.50-7.48(m,7H),7.15-7.12(m,2H),2.54(s,3H),2.37(s,3H);
13C NMR(100MHz,CDCl3,ppm):δ168.2,164.1,164.0,139.5,138.3,137.5,136.5,135.9,132.2,130.8,130.7,129.8,129.0,128.6,128.5,127.3,127.0,114.32,21.3,20.9;
MS(EI,70eV)m/z(%)=336[M+],259,217,168,129,104,77;
HRMS ESI(m/z):calcd for C24H20N2Na(M+Na)+:359.1519,found:359.1523.
根据以上数据推断所得产物的结构式如下式所示:
实施例8
在螺口试管中加入0.20毫摩尔4-嘧啶甲脒盐酸盐、0.25毫摩尔苯乙炔、0.25毫摩尔2,4-二甲基苯甲醛、0.4毫摩尔叔丁醇钾,1毫升二甲基亚砜,氮气保护条件下,在120℃搅拌反应12小时,停止加热及搅拌,冷却至室温。乙酸乙酯萃取反应液,减压旋蒸去除溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为60:1的石油醚:乙酸乙酯混合溶剂,产率为56%。
本实施例所得化合物的氢谱图和碳谱图分别如图3和图4所示,其结构表征数据如下:
IR(KBr)3042,2922,2860,1730,1587,1516,1456,1368,1033,810,740,679cm-1;
1H NMR(400MHz,CDCl3,ppm):δ8.78-8.77(m,2H),8.47(d,J=8.0MHz,2H),8.23(d,J=8.0MHz,2H),7.77(s,1H),7.53-7.47(m,4H),7.15(d,J=8.0MHz,2H),2.53(s,3H),2.39(s,3H);
13C NMR(100MHz,CDCl3,ppm):δ168.5,164.4,162.0,159.4,145.5,139.9,136.9,136.5,135.2,132.3,131.1,129.8,129.0,127.3,127.0,122.3,115.6,21.3,20.8;
MS(EI,70eV)m/z(%)=337[M+],281,232,207,129,115,77;
HRMS ESI(m/z):calcd for C23H20N3(M+H)+:338.1652,found:338.1660.
根据以上数据推断所得产物的结构式如下式所示:
实施例9
在螺口试管中加入0.20毫摩尔乙脒盐酸盐、0.25毫摩尔苯乙炔、0.25毫摩尔2,4-二甲基苯甲醛、0.4毫摩尔叔丁醇钾,1毫升二甲基亚砜,氮气保护条件下,在120℃搅拌反应12小时,停止加热及搅拌,冷却至室温。乙酸乙酯萃取反应液,减压旋蒸去除溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为10:1的石油醚:乙酸乙酯混合溶剂,产率为62%。
本实施例所得化合物的氢谱图和碳谱图分别如图5和图6所示,其结构表征数据如下:
IR(KBr)2918,2854,1641,1575,1526,1452,1381,758,686cm-1;
1H NMR(400MHz,CDCl3,ppm):δ8.10-8.09(m,2H),7.59(s,1H),7.50-7.49(m,3H),7.38(d,J=8.0MHz,1H),7.12(d,J=8.0MHz,2H),2.85(s,3H),2.42(s,3H),2.37(s,3H);
13C NMR(100MHz,CDCl3,ppm):δ168.0,168.0,164.1,139.3,137.4,135.8,135.8,131.9,130.7,129.5,129.0,127.3,126.9,113.9,26.5,21.2,20.3;
MS(EI,70eV):m/z(%)=274[M+],232,197,129,102,77;
HRMS ESI(m/z):calcd for C19H19N2(M+H)+:275.1543,found:275.1544.
根据以上数据推断所得产物的结构式如下式所示:
实施例10
在螺口试管中加入0.20毫摩尔4-氟苯甲脒盐酸盐、0.25毫摩尔苯乙炔、0.25毫摩尔2,4-二甲基苯甲醛、0.4毫摩尔叔丁醇钾,1毫升二甲基亚砜,氮气保护条件下,在120℃搅拌反应12小时,停止加热及搅拌,冷却至室温。乙酸乙酯萃取反应液,减压旋蒸去除溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为500:1的石油醚:乙酸乙酯混合溶剂,产率为64%。
本实施例所得化合物的氢谱图和碳谱图分别如图7和图8所示,其结构表征数据如下:
IR(KBr)2920,2855,1638,1458,1383,1028,953,756,679cm-1;
1H NMR(400MHz,CDCl3,ppm):δ8.69-8.66(m,2H),8.27-8.24(m,2H),7.72(s,1H),7.56-7.50(m,4H),7.21-7.17(m,4H),2.56(s,3H),2.42(s,3H);
13C NMR(100MHz,CDCl3,ppm):δ168.2,164.1,163.5,163.1,139.6,137.4,136.4,135.7,134.4(d,J=3.0Hz),132.1,130.8,130.6(d,J=8.0Hz),129.7,129.0,127.3,126.9,115.4(d,J=22.0Hz),114.2,21.3,20.7;
MS(EI,70eV):m/z(%)=354[M+],277,232,129,102,77;
HRMS ESI(m/z):calcd for C24H19FN3Na(M+H)+:377.1424,found:377.1422.
根据以上数据推断所得产物的结构式如下式所示:
实施例11
在螺口试管中加入0.20毫摩尔3-甲基苯甲脒盐酸盐、0.25毫摩尔苯乙炔、0.25毫摩尔2,4-二甲基苯甲醛、0.4毫摩尔叔丁醇钾,1毫升二甲基亚砜,氮气保护条件下,在120℃搅拌反应12小时,停止加热及搅拌,冷却至室温。乙酸乙酯萃取反应液,减压旋蒸去除溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为200:1的石油醚:乙酸乙酯混合溶剂,产率为59%。
本实施例所得化合物的氢谱图和碳谱图分别如图9和图10所示,其结构表征数据如下:
IR(KBr)3043,2923,2860,1572,1445,1361,1229,1085,818,766,694cm-1;
1H NMR(400MHz,CDCl3,ppm):δ8.49(s,2H),8.28(d,2H,J=4.0MHz),7.72(s,1H),7.54(d,J=12.0MHz,4H),7.44-7.40(m,1H),7.33(d,J=8.0MHz,1H),7.19(d,J=12.0MHz,2H),2.57(s,3H),2.50(s,3H),2.43(s,3H);
13C NMR(100MHz,CDCl3,ppm):δ168.2,164.2,164.1,139.5,138.2,138.0,137.6,136.4,135.9,132.1,131.4,130.7,129.7,129.1,128.9,128.4,127.3,126.9,125.7,114.3,21.6,21.3,20.7;
MS(EI,70eV):m/z(%)=350[M+],273,189,129,91,77;
HRMS ESI(m/z):calcd for C25H22N2Na(M+Na)+:373.1675,found:373.1676.
根据以上数据推断所得产物的结构式如下式所示:
实施例12
在螺口试管中加入0.20毫摩尔环丙甲脒盐酸盐、0.25毫摩尔苯乙炔、0.25毫摩尔2,4-二甲基苯甲醛、0.4毫摩尔叔丁醇钾,1毫升二甲基亚砜,氮气保护条件下,在120℃搅拌反应12小时,停止加热及搅拌,冷却至室温。乙酸乙酯萃取反应液,减压旋蒸去除溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为60:1的石油醚:乙酸乙酯混合溶剂,产率为70%。
本实施例所得化合物的氢谱图和碳谱图分别如图11和图12所示,其结构表征数据如下:
IR(KBr)2924,2859,1574,1450,1367,1281,1034,930,816,768,691cm-1;
1H NMR(400MHz,CDCl3,ppm):δ8.13-8.11(m,2H),7.56(s,1H),7.52-7.49(m,3H),7.41(d,J=8.0MHz,1H),7.13(d,J=8.0MHz,2H),2.46(s,3H),2.39(s,3H),1.31-1.28(m,2H),1.12-1.08(m,2H);
13C NMR(100MHz,CDCl3,ppm):δ171.7,167.7,163.6,136.2,135.9,132.0,130.6,129.5,128.9,127.2,126.8,113.4,21.2,20.6,18.5,10.6;
MS(EI,70eV):m/z(%)=300[M+],223,182,142,101,77;
HRMS ESI(m/z):calcd for C21H21N2(M+H)+:301.1699,found:301.1705.
根据以上数据推断所得产物的结构式如下式所示:
实施例13
在螺口试管中加入0.20毫摩尔苯甲脒盐酸盐、0.25毫摩尔4-戊基苯乙炔、0.25毫摩尔2,4-二甲基苯甲醛、0.4毫摩尔叔丁醇钾,1毫升二甲基亚砜,氮气保护条件下,在120℃搅拌反应12小时,停止加热及搅拌,冷却至室温。乙酸乙酯萃取反应液,减压旋蒸去除溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为300:1的石油醚:乙酸乙酯混合溶剂,产率为64%。
本实施例所得化合物的氢谱图和碳谱图分别如图13和图14所示,其结构表征数据如下:
IR(KBr)3031,2925,2858,1568,1514,1455,1356,816,753,690cm-1;
1H NMR(400MHz,CDCl3,ppm):δ8.73(d,J=8.0MHz,2H),8.23(d,J=8.0MHz,2H),7.73(s,1H),7.55(d,J=8.0MHz,4H),7.39(d,J=8.0MHz,2H),7.21(d,J=8.0MHz,2H),2.75-2.72(m,2H),2.60(s,3H),2.45(s,3H),1.74-1.70(m,2H),1.40(s,4H),0.98-0.95(m,3H);
13C NMR(100MHz,CDCl3,ppm):δ168.1,164.1,164.0,146.2,139.4,138.4,136.5,136.0,135.0,132.1,130.5,129.7,129.1,128.5,128.5,127.2,126.9,114.0,35.9,31.5,31.1,22.6,21.3,20.8,14.1;
MS(EI,70eV):m/z(%)=406[M+],348,259,217,154,115,91,77;
HRMS ESI(m/z):calcd for C29H31N2(M+H)+:407.2482,found:407.2483.
根据以上数据推断所得产物的结构式如下式所示:
实施例14
在螺口试管中加入0.20毫摩尔苯甲脒盐酸盐、0.25毫摩尔4-甲基苯乙炔、0.25毫摩尔2,4-二甲基苯甲醛、0.4毫摩尔叔丁醇钾,1毫升二甲基亚砜,氮气保护条件下,在120℃搅拌反应12小时,停止加热及搅拌,冷却至室温。乙酸乙酯萃取反应液,减压旋蒸去除溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为500:1的石油醚:乙酸乙酯混合溶剂,产率为63%。
本实施例所得化合物的氢谱图和碳谱图分别如图15和图16所示,其结构表征数据如下:
IR(KBr)3031,2923,2862,1572,1517,1448,1362,1180,819,760,696cm-1;
1H NMR(400MHz,CDCl3,ppm):δ8.73(d,J=4.0MHz,2H),8.21(d,J=8.0MHz,2H),7.72(s,1H),7.58-7.54(m,4H),7.38(d,J=8.0MHz,2H),7.21(d,J=12.0MHz,2H),2.61(d,J=8.0MHz,3H),2.48(s,3H),2.45(s,3H);
13C NMR(100MHz,CDCl3,ppm):δ168.1,164.0,164.0,141.1,139.4,138.4,136.5,136.0,134.7,132.1,130.5,129.8,129.7,128.5,128.5,127.2,126.9,114.0,21.5,21.3,20.8;
MS(EI,70eV):m/z(%)=350[M+],259,175,129,104,77;
HRMS ESI(m/z):calcd for C25H23N2(M+H)+:351.1856,found:351.1864.
根据以上数据推断所得产物的结构式如下式所示:
实施例15
在螺口试管中加入0.20毫摩尔苯甲脒盐酸盐、0.25毫摩尔3-氯苯乙炔、0.25毫摩尔2,4-二甲基苯甲醛、0.4毫摩尔叔丁醇钾,1毫升二甲基亚砜,氮气保护条件下,在120℃搅拌反应12小时,停止加热及搅拌,冷却至室温。乙酸乙酯萃取反应液,减压旋蒸去除溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为100:1的石油醚:乙酸乙酯混合溶剂,产率为59%。
本实施例所得化合物的氢谱图和碳谱图分别如图17和图18所示,其结构表征数据如下:
IR(KBr)3066,2922,2854,1567,1356,1231,815,760,697cm-1;
1H NMR(400MHz,CDCl3,ppm):δ8.67-8.65(m,2H),8.28(s,1H),8.12(d,J=8.0Hz,1H),7.69(s,1H),7.54-7.46(m,6H),7.19(d,J=12.0MHz,2H),2.57(s,3H),2.42(s,3H);
13C NMR(100MHz,CDCl3,ppm):δ168.5,164.1,162.7,139.7,139.4,138.0,136.5,135.6,135.1,132.2,130.8,130.7,130.2,129.7,128.5,127.4,126.9,125.3,114.3,21.3,20.8;
MS(EI,70eV):m/z(%)=370[M+],259,202,104,63;
HRMS ESI(m/z):calcd for C24H20ClN2(M+H)+:371.1310,found:371.1311.
根据以上数据推断所得产物的结构式如下式所示:
实施例16
在螺口试管中加入0.20毫摩尔苯甲脒盐酸盐、0.25毫摩尔4-氯苯乙炔、0.25毫摩尔2,4-二甲基苯甲醛、0.4毫摩尔叔丁醇钾,1毫升二甲基亚砜,氮气保护条件下,在120℃搅拌反应12小时,停止加热及搅拌,冷却至室温。乙酸乙酯萃取反应液,减压旋蒸去除溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为500:1的石油醚:乙酸乙酯混合溶剂,产率为57%。
本实施例所得化合物的氢谱图和碳谱图分别如图19和图20所示,其结构表征数据如下:
IR(KBr)3063,2921,2853,1571,1448,1359,1091,820,756,694cm-1;
1H NMR(400MHz,CDCl3,ppm):δ8.67-8.74(m,2H),8.22(d,J=8.0MHz,2H),7.69(s,1H),7.53-7.50(m,6H),7.18(t,J=8.0MHz,2H),2.57(s,3H),2.42(s,3H);
13C NMR(100MHz,CDCl3,ppm):δ168.4,164.1,162.9,139.6,138.1,137.0,136.5,135.9,135.7,132.2,130.7,129.7,129.2,128.5,128.5,128.5,126.9,114.0,21.3,20.8;
MS(EI,70eV):m/z(%)=370[M+],307,259,207,104,77;
HRMS ESI(m/z):calcd for C24H20N2Cl(M+H)+:371.1310,found:371.1308.
根据以上数据推断所得产物的结构式如下式所示:
实施例17
在螺口试管中加入0.20毫摩尔苯甲脒盐酸盐、0.25毫摩尔2-甲氧基苯乙炔、0.25毫摩尔2,4-二甲基苯甲醛、0.4毫摩尔叔丁醇钾,1毫升二甲基亚砜,氮气保护条件下,在120℃搅拌反应12小时,停止加热及搅拌,冷却至室温。乙酸乙酯萃取反应液,减压旋蒸去除溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为60:1的石油醚:乙酸乙酯混合溶剂,产率为57%。
本实施例所得化合物的氢谱图和碳谱图分别如图21和图22所示,其结构表征数据如下:
IR(KBr)3060,2929,2845,1568,1362,1246,1027,819,755,697cm-1;
1H NMR(400MHz,CDCl3,ppm):δ8.75(t,J=4.0MHz,2H),8.38-8.36(m,1H),8.09(s,1H),7.65-7.50(m,5H),7.28-7.23(m,3H),7.09(d,J=8.0MHz,1H),3.96(s,3H),2.67(s,3H),2.48(s,3H);
13C NMR(100MHz,CDCl3,ppm):δ166.8,163.8,162.6,158.2,139.3,138.6,136.5,136.3,132.1,131.6,131.5,130.4,130.0,128.5,128.4,127.0,126.9,121.2,119.5,111.6,55.7,21.3,20.8;
MS(EI,70eV):m/z(%)=366[M+],259,207,154,104,63;
HRMS ESI(m/z):calcd for C25H23N2O(M+H)+:367.1805,found:367.1811.
根据以上数据推断所得产物的结构式如下式所示:
实施例18
在螺口试管中加入0.20毫摩尔苯甲脒盐酸盐、0.25毫摩尔苯乙炔、0.25毫摩尔4-甲氧基苯甲醛、0.4毫摩尔叔丁醇钾,1毫升二甲基亚砜,氮气保护条件下,在120℃搅拌反应12小时,停止加热及搅拌,冷却至室温。乙酸乙酯萃取反应液,减压旋蒸去除溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为30:1的石油醚:乙酸乙酯混合溶剂,产率为65%。
本实施例所得化合物的氢谱图和碳谱图分别如图23和图24所示,其结构表征数据如下:
IR(KBr)3061,2924,2841,1571,1520,1363,1247,1175,1029,833,754,69cm-1;
1H NMR(400MHz,CDCl3,ppm):δ8.76-8.72(m,2H),8.30-8.26(m,4H),7.93(s,1H),7.58-7.54(m,6H),7.07(d,J=12.0MHz,2H),3.89(s,3H);
13C NMR(100MHz,CDCl3,ppm):δ164.5,164.4,164.2,162.0,138.4,137.7,130.7,130.5,130.0,128.9,128.8,128.5,128.4,127.3,114.3,109.4,55.4;
MS(EI,70eV):m/z(%)=338[M+],235,207,132,102,63;
HRMS ESI(m/z):calcd for C23H19N2O(M+H)+:339.1492,found:339.1494.
根据以上数据推断所得产物的结构式如下式所示:
实施例19
在螺口试管中加入0.20毫摩尔苯甲脒盐酸盐、0.25毫摩尔苯乙炔、0.25毫摩尔4-甲基苯甲醛、0.4毫摩尔叔丁醇钾,1毫升二甲基亚砜,氮气保护条件下,在120℃搅拌反应12小时,停止加热及搅拌,冷却至室温。乙酸乙酯萃取反应液,减压旋蒸去除溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为300:1的石油醚:乙酸乙酯混合溶剂,产率为62%。
本实施例所得化合物的氢谱图和碳谱图分别如图25和图26所示,其结构表征数据如下:
IR(KBr)3045,2922,2856,1576,1521,1362,1028,824,750,688cm-1;
1H NMR(400MHz,CDCl3,ppm):δ8.76(t,J=4.0MHz,2H),8.30(t,J=4.0MHz,2H),8.21(d,J=8.0MHz,2H),7.98(s,1H),7.59-7.54(m,6H),7.37(d,J=8.0MHz,2H),2.47(s,3H);
13C NMR(100MHz,CDCl3,ppm):δ164.7,164.6,164.5,141.1,138.3,137.7,134.8,130.7,130.6,129.7,128.9,128.5,128.4,127.3,127.2,109.9,21.5;
MS(EI,70eV):m/z(%)=322[M+],219,204,161,102,77;
HRMS ESI(m/z):calcd for C23H19N2(M+H)+:323.1543,found:323.1549.
根据以上数据推断所得产物的结构式如下式所示:
实施例20
在螺口试管中加入0.20毫摩尔苯甲脒盐酸盐、0.25毫摩尔苯乙炔、0.25毫摩尔苯甲醛、0.4毫摩尔叔丁醇钾,1毫升二甲基亚砜,氮气保护条件下,在120℃搅拌反应12小时,停止加热及搅拌,冷却至室温。乙酸乙酯萃取反应液,减压旋蒸去除溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为300:1的石油醚:乙酸乙酯混合溶剂,产率为68%。
本实施例所得化合物的氢谱图和碳谱图分别如图27和图28所示,其结构表征数据如下:
IR(KBr)3053,2920,2851,1572,1525,1444,1359,1236,856,737,681cm-1;
1H NMR(400MHz,CDCl3,ppm):δ8.79-8.78(m,2H),8.33-8.30(m,4H),8.01(s,1H),7.61-7.54(m,9H);
13C NMR(100MHz,CDCl3,ppm):δ164.8,164.5,138.3,137.6,130.8,130.7,128.9,128.6,128.5,127.3,110.3;
MS(EI,70eV):m/z(%)=308[M+],154,128,76;
HRMS ESI(m/z):calcd for C22H17N2(M+H)+:309.1386,found:309.1389.
根据以上数据推断所得产物的结构式如下式所示:
实施例21
在螺口试管中加入0.20毫摩尔苯甲脒盐酸盐、0.25毫摩尔苯乙炔、0.25毫摩尔呋喃甲醛、0.4毫摩尔叔丁醇钾,1毫升二甲基亚砜,氮气保护条件下,在120℃搅拌反应12小时,停止加热及搅拌,冷却至室温。乙酸乙酯萃取反应液,减压旋蒸去除溶剂,再通过柱层析分离纯化,得到目标产物,所用的柱层析洗脱液为体积比为30:1的石油醚:乙酸乙酯混合溶剂,产率为62%。
本实施例所得化合物的氢谱图和碳谱图分别如图29和图30所示,其结构表征数据如下:
IR(KBr)3055,2923,2853,1601,1551,1485,1359,822,743,689cm-1;
1H NMR(400MHz,CDCl3,ppm):δ8.69-8.67(m,2H),8.31-8.29(m,2H),7.95(s,1H),7.65(s,1H),7.59-7.52(m,6H),7.47(d,J=4.0MHz,1H),6.64-6.63(m,1H);
13C NMR(100MHz,CDCl3,ppm):δ164.6,164.5,156.4,152.7,144.8,138.0,137.4,130.9,130.7,128.9,128.9,128.4,127.3,112.5,112.1,108.0;
MS(EI,70eV):m/z(%)=298[M+],270,195,167,102,92,63;
HRMS ESI(m/z):calcd for C20H15N2O(M+H)+:299.1179,found:299.1179.
根据以上数据推断所得产物的结构式如下式所示:
本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!