一种基于主链型苯并噁嗪制备聚苯并噁唑的方法与流程
本发明属于热固性树脂及其制备技术领域,具体涉及主链型苯并噁嗪(中间体)、高性能聚苯并噁唑及其制备方法。
技术背景
聚苯并噁唑及其衍生物是分子结构中含有氧原子和氮原子的苯并杂环类有机高分子。此类高分子化合物具有耐高温、高刚性、抗菌、防腐等诸多优异的性能,在航天航空、军工、医药、农药等领域有着广泛应用。特别是近年来随着我国航天航空事业的快速发展,高性能材料的需求量日益增加,苯并噁唑及其衍生物正逐渐成在航天航空、军工领域被研究较多的一类高性能聚合物,因而关于聚苯并噁唑及其衍生物的合成方法也越来越受到人们广泛关注。
在目前已报道的相关专利中,关于聚苯并噁唑及其衍生物的合成方法有以下几种。如申请号为200610155718.3的专利“5-氨基-6-羟基-2-(对羟基苯基)苯并噁唑盐、合成及应用”(公开号CN101016275A),公布了聚对苯撑苯并二噁唑的制备,主要步骤有以多聚磷酸为溶剂、五氧化二磷为脱水剂,5-氨基-6-羟基-2-(对羟基苯基)苯并噁唑羧铵盐为反应物在N2保护下90-230℃阶梯升温自缩聚制备;如申请号为200810040907.5的专利“聚苯并噁唑的制备方法”(公开号CN101323664A),公布了一种聚苯并噁唑的制备方法,主要步骤有在一定量五氧化二磷的多聚磷酸溶剂中,在惰性气体的保护下,配制出4,6-二氨基间苯二酚/对苯甲二酸复合盐的多聚磷酸溶液通过双螺杆进行反应性挤出聚合,制备得到聚苯并噁唑;如申请号为201310552551.4的专利“一种苯并噁唑化合物的合成方法”(公开号CN103554050A),公布了一种苯并噁唑化合物的合成方法,主要步骤有以邻氨基苯酚、1,3-二羰基化合物、质子酸和铜盐催化剂油浴反应,反应结束后,除去溶剂,使用石油醚/乙酸乙酯为洗脱剂,硅胶柱分离,制得苯并噁唑类化合物;如申请号为201511031122.8的专利“聚苯并噁唑的制备方法”(公开号CN105524011A),公布了一种钯催化的2-(2-羟基苯基)苯并噁唑的合成方法,主要步骤有水杨醛和2-氨基苯酚进行缩合反应后,将该中间体、Pd(OAc)2和Cs2CO3溶于有机溶剂后氧化成环,接着反应完全后进一步洗脱除去金属钯、其它杂质离子和溶剂,最终得到化合物聚苯并噁唑。
这些专利,通过在五氧化二磷的多聚磷酸溶剂或者在含有质子酸和金属催化剂的酸性条件下,均成功制备出苯并噁唑及其衍生物,但是进一步分析此类合成方案发现存在以下缺点:(1)在合成反应过程中磷酸溶剂无法完全除尽,导致产物在高湿、高温条件下长期使用过程中苯并噁唑环易开环分解;(2)在酸性条件下制备产物对加工设备腐蚀较大,且反应流程复杂;(3)上述制备合成中使用了重金属催化剂,不仅催化剂昂贵且易造成环境污染。然而关于通过点击化学反应合成中间体(主链型聚苯并噁嗪)从而制备聚苯并噁唑并未提及。最近发展的点击化学反应经常被应用在高分子合成中,用来修饰或合成新型高分子结构,但目前的专利中并未出现通过苯并噁嗪单体与叠氮化合物进行点击化学反应,在室温条件下制备主链型苯并噁嗪中间体从而制备聚苯并噁唑的报道。此外,截止目前还未有报道涉及具有特殊基团的主链型苯并噁嗪热固化后通过热环化得到具有高刚性的聚苯并噁唑热固性树脂材料。
技术实现要素:
本发明的目的是针对已有技术存在的不足,提供一种基于主链型苯并噁嗪制备高刚性聚苯并噁唑的制备方法。
本发明的目的是通过以下技术方案来实现的:
一种聚苯并噁唑,分子式为:
其中,为以下之一:
-R2-为以下之一:
所述聚苯并噁唑的热失重5%时,温度为500-530℃;惰性气体氛围800℃时,残炭率为70-80%。
所述聚苯并噁唑是基于主链型苯并噁嗪制备得到的。
一种基于主链型苯并噁嗪制备高刚性聚苯并噁唑的方法,具体包括如下步骤:
(1)含邻位酰胺基团的二酚的合成:
先将2-胺基苯酚与间/对苯二甲酰氯混合,以LiCl为催化剂,加入到NMP溶剂体系中,反应体系中充满惰性保护气体,在室温下反应时间24h停止反应后将体系用去离子水洗涤、过滤、干燥,得到含邻位酰胺基团的二酚;
反应方程式为:
步骤(1)中,所述的2-胺基苯酚与间/对苯二甲酰氯摩尔比为1:2~1:2.5,优选为1:2.2;
步骤(1)中,所述的惰性保护气体为氮气或氩气;
(2)含有酰胺基、炔基的苯并噁嗪单体的合成:
将步骤(1)制备的含邻位酰胺基团的二酚和含炔基的苯胺、多聚甲醛在二甲苯溶剂中混合,然后升温至120℃,反应时间6h,停止反应后,用碱液洗涤并挥发溶剂得到固体,再经烘干得到产物,即为含有酰胺基、炔基的苯并噁嗪单体;
反应方程式为:
其中,为以下之一:
步骤(2)中,所述的含邻位酰胺基团的二酚和含炔基的苯胺、多聚甲醛按摩尔比为1:2:4~1:2:5,优选为1:2:4.4。
步骤(2)中,所述的碱液为浓度5%~10%的氢氧化钠水溶液。
(3)主链型苯并噁嗪的制备:
将步骤(2)得到的含有酰胺基、炔基的苯并噁嗪单体与双官能叠氮化合物混合后,加入到干燥处理的DMF溶剂体系中,在室温下进行点击化学反应12h,得到目标产物之一主链型苯并噁嗪;
反应方程式为:
其中,-R2-为以下之一:
步骤(3)中,所述的苯并噁嗪单体与双官能叠氮化合物的摩尔比为1:1~1:1.5,优选为1:1.2;
(4)通过主链型苯并噁嗪制备高性能聚苯并噁唑:
将步骤(3)得到的主链型苯并噁嗪溶于有机溶剂中配备成溶液,再将溶液浇筑于涂有脱模剂的模具中,首先在100℃条件下缓慢热处理24h除去溶剂,之后在200℃固化2h得到苯并噁嗪树脂,再经300℃热环化处理得到具有高刚性苯并噁唑热固性树脂材料。
步骤(4)中,所述的有机溶剂为二甲基亚砜、二甲基甲酰胺、二甲基乙酰胺中的一种或混合物;
所述聚苯并噁唑可作为基体材料广泛的应用于航空航天、电子封装领域。
与现有技术相比,本发明的优点在于:
(1)利用含有邻位酰胺基的主链型苯并噁嗪的热固化产生酚羟基,再将酚羟基与其邻位酰胺基进行苯并噁唑热环化来制备高刚性苯并噁唑热固性树脂材料,避免使用了环境有害的强质子酸作为反应溶剂;
(2)制备主链型聚苯并噁唑操作步骤简单,原料易得,环境友好且易于工业化生产。
附图说明
图1实施例1得到的聚苯并噁唑的红外光谱图;
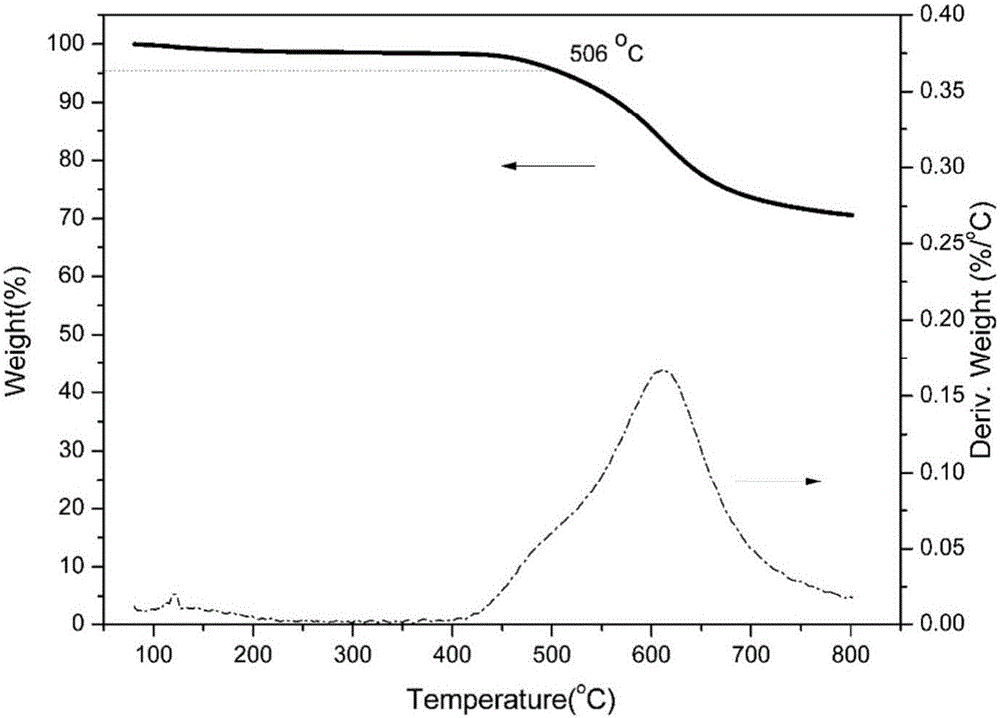
图2实施例1得到的聚苯并噁唑的TGA谱图。
具体实施方式
下面通过结合实例对本发明进行具体描述说明。有必要指出的是:以下实例只用于对本发明进行更详细的说明,并不局限本发明的保护范围。本技术领域的普通专业人员阅读本发明之后,在没有做出创造性劳动的前提下所获得的所有其它实施实例,都属于本发明保护的范围。
实施例1
具体步骤为:
(1)将10.000g的间苯二甲酰氯、10.751g的2-氨基苯酚、3.450g LiCl和80.000mL的N-甲基吡咯烷酮(NMP)加入到装有冷凝管的反应瓶中,反应体系中充满氮气,刚开始在冰浴条件下然后在室温下反应,反应24h后结束。将产物缓慢倒入去离子水中,静置出现沉淀后进行抽滤,水洗至中性后,将产物放入真空烘箱烘干,得到产物纯净的含邻位酰胺基团的二酚14.530g,收率85%。反应方程如下:
(2)称取由上一步反应得到的二酚5.000g,以及多聚甲醛1.793g,3-乙炔基苯胺3.363g以及二甲苯60mL分别加入到装有搅拌器、温度计、及冷凝管的反应瓶中,逐步升温至120℃搅拌6h后。反应结束后用低浓度(5%-10%)的氢氧化钠洗涤并过滤除去未溶解的杂质,旋蒸回收溶剂后得到固体7.831g,收率86%。反应方程式如下:
(3)称取上一步得到的含有酰胺、炔基苯并噁嗪单体5.000g,然后和叠氮化合物3.746g在室温下进行点击化学反应,反应12h,反应结束后,将反应液倒入去离子水中,沉淀、过滤、干燥得到产物7.171g,收率82%,反应方程式如下:
(4)将上一步得到的主链型苯并噁嗪溶于DMSO中配备成一定浓度的溶液,再将溶液浇筑于涂有脱模剂的模具中。首先在100℃条件下缓慢热处理24h除去溶剂,之后在200℃固化2h得到苯并噁嗪树脂,再经300℃苯并噁唑热环化处理得到高刚性的聚苯并噁唑材料。得到的聚苯并噁唑产物结构如下:
该产物的傅里叶红外变换光谱图和热失重曲线图见附图1和附图2。
附图1为红外光谱图,其中1623cm-1处为苯并噁唑环的特征吸收峰。
图2为热失重曲线图,可以看出,聚苯并噁唑热失重5%时的温度为506℃,800℃时的残炭率为71%。
实施例2
将实施例1中的第二步中的反应物3-乙炔基苯胺替换为3-乙炔基-4-甲基苯胺,反应物的量做相应的改变,其它操作步骤同实施例1中的步骤。
在第二步反应中,反应物的量更改为:由上一步反应得到的二酚5.000g,以及多聚甲醛1.793g,3-乙炔基-5-甲基苯胺3.766g。
其中得到的主链型聚苯并噁嗪具体化学结构为:
其中得到的聚苯并噁唑具体化学结构为:
本实施例得到的聚苯并噁唑其热失重5%时的温度为501℃,800℃时的残炭率为70%。
实施例3
将实施例1中的叠氮化合物替换为反应物的量做相应的改变,其它操作步骤同实施例1中的步骤。
在第三步反应中,反应物的量更改为:含有酰胺基、炔基苯并噁嗪单体5.000g,叠氮化合物3.968g。
其中得到的主链型聚苯并噁嗪具体化学结构为:
其中得到的主链型聚苯并噁唑具体化学结构为:
本实施例得到的聚苯并噁唑其热失重5%时的温度为515℃,800℃时的残炭率为76%。
- 还没有人留言评论。精彩留言会获得点赞!