一种高对应选择性1,1‑二芳基硼化烷烃类化合物及其制备方法与流程
本发明属于有机合成化学技术领域,具体涉及一种高对应选择性1,1-二芳基硼化烷烃类化合物及其制备方法。
背景技术:
手性不对称双芳基或芳烷基的三级碳结构普遍存在于大量生物活性物种或药物分子中,例如,花青醛、(R)-托特罗定、足叶草霉素、丁苯吗啡等,使其在药物化学、生物化学和材料科学上都起着十分重要的作用。因此,发展高对应选择性的1,1-二取代烷烃类化合物对有机合成化学具有重要的研究价值(Schmidt,F.;Stemmler,R.T.;Rudolph,J.;Bolm,C.Chem.Soc.Rew.2006,35,454.)。
近年来,过渡金属Rh、Co、Ir、Fe、Cu催化烯烃的氢硼化反应备受关注,大量合成对应选择性烷烃的文献被报道(Smith,S.M.;Takacs,J.M.J.Am.Chem.Soc.2010,132,1740.)。但是,这类文献主要阐述了单取代烯烃以及1,2-二取代烯烃的不对称反应,对于1,1-二芳基烯烃的对应选择性不是很乐观。经过调查研究,主要原因是区分前手性底物的两个相似平面具有很大挑战,以至于很难合成高对应选择性的1,1-二芳基烷烃类化合物。例如,过渡金属Co催化烯烃氢硼化的两个例子中,1,1-二芳基烷烃仅有8%和54%的低对应选择性的结果(Zhang,L.;Zuo,Z;Wan,X.;Huang,Z.J.Am.Chem.Soc.2014)。
随着全球生态环境的急剧恶化,如何实现可持续发展已成为人类面临的重大问题,以从源头上消除污染、节省资源为核心的绿色化学研究已经成为解决日益严峻的生态环境问题的强有力手段。Cu催化1,1-二芳基烯烃不对称氢硼化反应就具有环境友好、价格低廉、实用高效等优点。到目前为止,具有高对应选择性硼化的1,1-二芳基烷烃类化合物的反应还未见文献报道。
技术实现要素:
本发明提供一种高对应选择性1,1-二芳基硼化烷烃类化合物及其制备方法,该化合物具有高对应选择性。
本发明首先提供一种高对应选择性1,1-二芳基硼化烷烃类化合物,该化合物的结构通式如式Ⅰ所示:
式Ⅰ中,R1为芳基、烷基或氨基,R2为芳基、烷基或杂芳基。
优选的是,所述的R1为苯基、甲基、乙基、异丙基、烷氧基或N,N-二甲基。
优选的是,R2为苯基、吲哚、噻吩或烷氧基。
优选的是,所述的高对应选择性1,1-二芳基硼化烷烃类化合物具有2a-2s所示的结构:
本发明还提供一种高对应选择性1,1-二芳基硼化烷烃类化合物的制备方法,该方法包括:
步骤一:将二芳基甲酮、Wittig试剂和正丁基锂在极性溶剂中反应,得到1,1-二芳基乙烯化合物;
步骤二:在反应装置中加入催化剂、配体((2S,4S)-(-)-2,4-双(二苯基磷)戊烷)、抗衡离子和碱,然后再加入步骤一得到的1,1-二芳基乙烯化合物和联硼酸频那醇酯反应,得到高对应选择性1,1-二芳基硼化烷烃类化合物。
优选的是,所述的二芳基甲酮、Wittig试剂与正丁基锂的摩尔比为1:(1.3-2):(1.3-2)。
优选的是,所述的二芳基甲酮为2-甲氧基二苯甲酮、2-甲基二苯甲酮、2-异丙基二苯甲酮、2-苯基二苯甲酮或1-萘基苯甲酮。
优选的是,所述的Wittig试剂为甲基三苯基溴化膦。
优选的是,所述的抗衡离子为(四(3,5-二(三氟甲基)苯基)硼酸钠;碱为叔丁醇钠。
优选的是,所述的1,1-二芳基乙烯化合物和联硼酸频那醇酯的摩尔比为1:(1.2-1.5)。
本发明的有益效果
本发明首先提供一种高对应选择性1,1-二芳基硼化烷烃类化合物,该化合物的结构通式如式Ⅰ所示,该1,1-二芳基硼化烷烃类化合物,是在碱的催化下,铜盐与碱生成叔丁氧铜中间体,之后与联硼酸频那醇酯生成铜硼物种插入碳碳双键,在醇的作用下得到氢硼化的产物。本发明的1,1-二芳基硼化烷烃类化合物具有高对应选择性,通过高效液相色谱实验证明其ee值可达90%以上。
本发明还提供一种高对应选择性1,1-二芳基硼化烷烃类化合物的制备方法,该方法原料试剂易得,条件温和,反应体系绿色环保,产物易分离纯化、操作简单,适用于合成多种高对应选择性的1,1-二芳基烷烃类化合物,可进行大规模工业生产。
附图说明
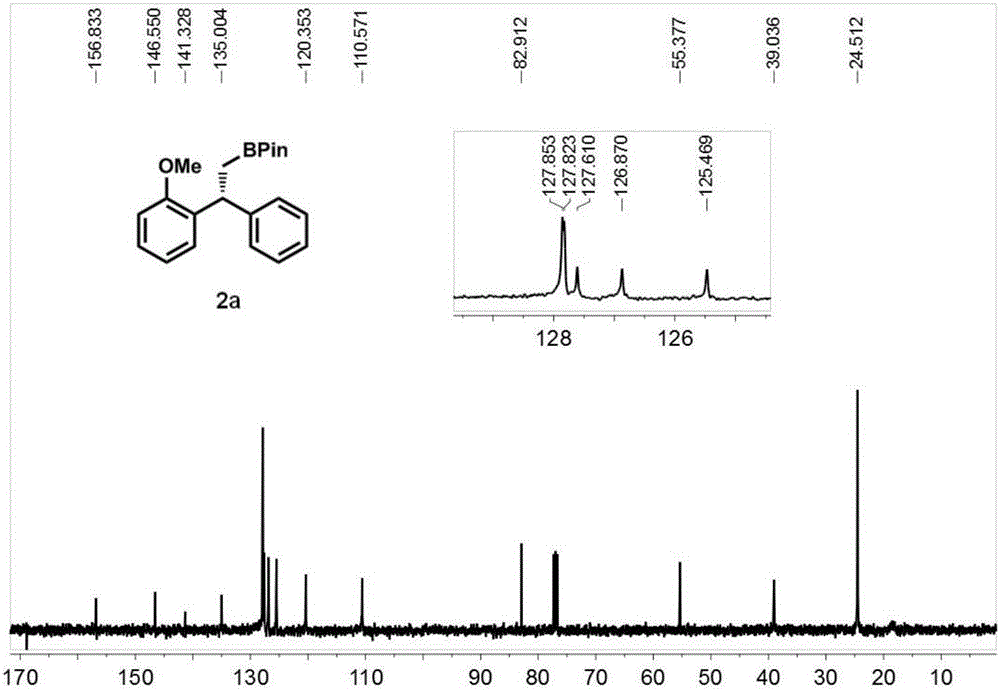
图1为实施例1制备得到的1,1-二芳基硼化烷烃类化合物2a的1H-NMR的核磁共振谱;
图2为实施例1制备得到的1,1-二芳基硼化烷烃类化合物2a的13C-NMR的核磁共振谱;
图3为实施例2制备得到的1,1-二芳基硼化烷烃类化合物2b的1H-NMR的核磁共振谱;
图4为实施例2制备得到的1,1-二芳基硼化烷烃类化合物2b的13C-NMR的核磁共振谱;
图5为实施例1制备得到的1,1-二芳基硼化烷烃类化合物2a的高效液相色谱图;
图6为实施例2制备得到的1,1-二芳基硼化烷烃类化合物2b的高效液相色谱图。
具体实施方式
本发明首先提供一种高对应选择性1,1-二芳基硼化烷烃类化合物,该化合物的结构通式如式Ⅰ所示:
式Ⅰ中,R1为芳基、烷基或氨基,优选为苯基、甲基、乙基、异丙基、烷氧基或N,N-二甲基;R2为芳基、烷基或杂芳基,优选为苯基、吲哚、噻吩或烷氧基。
按照本发明,所述的高对应选择性1,1-二芳基硼化烷烃类化合物优选具有2a-2s所示的结构:
本发明还提供一种高对应选择性1,1-二芳基硼化烷烃类化合物的制备方法,该方法包括:
步骤一:将二芳基甲酮、Wittig试剂和正丁基锂在极性溶剂中反应,得到1,1-二芳基乙烯化合物;
步骤二:在反应装置中加入催化剂、配体((2S,4S)-(-)-2,4-双(二苯基磷)戊烷)、抗衡离子和碱,然后再加入步骤一得到的1,1-二芳基乙烯化合物和联硼酸频那醇酯反应,得到高对应选择性1,1-二芳基硼化烷烃类化合物。
按照本发明,在反应装置中加入Wittig试剂和极性溶剂,优选将反应装置降温至-78℃,然后滴加正丁基锂,滴加完毕后,优选在室温下反应30min,然后在反应装置中加入二芳基甲酮,优选在室温下反应24h,TLC检测底物消失,反应结束,将反应产物经过萃取、干燥抽滤,得到1,1-二芳基乙烯化合物;
按照本发明,所述的二芳基甲酮、Wittig试剂与正丁基锂的摩尔比优选为1:(1.3-2):(1.3-2);所述的二芳基甲酮优选为2-甲氧基二苯甲酮、2-甲基二苯甲酮、2-异丙基二苯甲酮、2-苯基二苯甲酮或1-萘基苯甲酮;所述极性溶剂优选为四氢呋喃、乙醚、1,4-二氧六环或甲基叔丁基醚;所述的Wittig试剂优选为甲基三苯基溴化膦。
按照本发明,氮气保护下,在反应装置中加入催化剂、配体、抗衡离子和碱,再加入极性溶剂,优选在室温下搅拌10min,然后向反应装置中加入联硼酸频那醇酯,优选在室温下搅拌10min,再加入上述1,1-二芳基乙烯化合物和反应溶剂,优选在室温下搅拌24h,并用TLC检测反应完成,将反应物经淬灭、萃取,合并有机相、干燥、抽滤,加圧蒸留除去有机溶剂,得到1,1-二芳基烷烃类化合物。
所述的催化剂优选为氯化亚铜,催化剂的用量优选为1,1-二芳基乙烯化合物摩尔量的5mol%;配体优选为(S,S)-BDPP((2S,4S)-(-)-2,4-双(二苯基磷)戊烷),配体用量优选为1,1-二芳基乙烯化合物摩尔量的5mol%;本发明选用(S,S)-BDPP作为反应配体,(S,S)-BDPP对于两个芳基识别是十分高效的,主要由于其中一个芳基的2号位置存在取代基团,使其中的一个芳环位阻增大。因此由于位阻效应,在配体识别时,能够有效区分两个芳基,得到具有高对应选择性的产物。然而,如果芳环上的取代基存在于位阻效应较弱的3,4号位置时,那么配体对于两个芳环的选择性就会较低(ee<20%)。
所述的抗衡离子优选为NaBARF(四(3,5-二(三氟甲基)苯基)硼酸钠,抗衡离子用量优选为1,1-二芳基乙烯化合物摩尔量的5mol%;碱优选为叔丁醇钠,碱的用量优选为1,1-二芳基乙烯化合物摩尔量的15mmol%。
按照本发明,所述的1,1-二芳基乙烯化合物和联硼酸频那醇酯的摩尔比优选为1:(1.2-1.5)。所述极性溶剂优选为甲苯、乙醚、四氢呋喃、甲基叔丁基醚或二氯甲烷;更优选为四氢呋喃。所述的反应溶剂为甲醇。
下面结合具体实施例对本发明做进一步详细的说明,实施例中涉及到的原料均为商购。
实施例1
1)1,1-二芳基乙烯1a的制备
向带有磁力搅拌装置的25mL圆底烧瓶中加入甲基三苯基溴化膦(2.6mmol)和四氢呋喃(5ml)。将反应移到-78℃,向体系中滴加正丁基锂(2.6mmol),滴加完毕后,室温反应半小时,再向体系加入二芳基甲酮(2mmol),室温搅拌24h,TLC检测底物消失,反应结束。将饱和氯化铵水溶液(10mL)倒入反应中,用二氯甲烷(3×10mL)萃取,合并有机相,无水硫酸钠干燥、抽滤,然后减压蒸馏除去有机溶剂,经过硅胶柱层析(洗脱液为V石油醚:V乙醚=20:1),得到无色油状液体,产物的结构经过NMR、MS证实为1a,收率为90%。
2)1,1-二芳基烷烃的制备
氮气保护下,向带有磁力搅拌装置的25mL圆底烧瓶中加入氯化亚铜(5mol%)、(S,S)-BDPP(5mol%)、NaBARF(5mol%)和叔丁醇钠(0.15mmol),再加入四氢呋喃(2ml),室温下搅拌10min。搅拌10min后,向体系中加入联硼酸频那醇酯(0.18mmol),继续搅拌10min,然后,加入1a(0.15mmol)和甲醇(0.3mmol)。将反应放在室温搅拌24h,并用TLC检测反应完成。反应用水(10ml)淬灭,二氯甲烷(3×10ml)萃取,合并有机相,用无水硫酸钠干燥、抽滤,加圧蒸留除去有机溶剂。最后经过硅胶柱层析,得到1,1-二芳基烷烃类化合物2a,产率为88%,ee值为84%。
图1为实施例1制备得到的1,1-二芳基硼化烷烃类化合物2a的1H-NMR的核磁共振谱;图2为实施例1制备得到的1,1-二芳基硼化烷烃类化合物2a的13C-NMR的核磁共振谱;图5为实施例1制备得到的1,1-二芳基硼化烷烃类化合物2a的高效液相色谱图;其中图a为反应配体为三苯基膦,且不加NarBARF,其它反应条件和实施例1相同,得到的1,1-二芳基烷烃类化合物的高效液相色谱图;图b为本发明实施例1制备得到的1,1-二芳基硼化烷烃类化合物2a的高效液相色谱图,具体数据如下:
1H NMR(400MHz,CDCl3)δ:7.29-7.18(m,5H),7.14-7.07(m,2H),6.89(t,J=7.6Hz,1H),6.78(d,J=8.0Hz,1H),4.70(t,J=8.8Hz,1H),3.75(s,3H),1.57(d,J=8.4Hz,2H),1.03(s,6H),1.02(s,6H);13C NMR(100MHz,CDCl3)δ:156.83,146.55,141.33,135.00,127.85,127.82,127.61,126.87,125.47,120.35,110.57,82.91,55.38,39.04,24.51.Mass Spectrometry:HRMS(ESI-TOF)(m/z):Calcd for C21H27BNaO3([M+Na]+):361.1949;found:361.1959.[α]D28=+17.2,(c=0.40,CHCl3).HPLC analysis(Chiralpak AD-H column,hexanes/i-PrOH=98/2,0.5ml/min,254nm,tR(minor)=10.4min,tR(major)=12.8min).
从图5可以看出,本发明制备的实施例1制备得到的1,1-二芳基硼化烷烃类化合物具有高对应选择性。
实施例2
1)1,1-二芳基乙烯1b的制备
氮气保护下,向带有磁力搅拌装置的25mL圆底烧瓶中加入甲基三苯基溴化膦(2.6mmol)和四氢呋喃(5ml)。将反应移到-78℃,向体系中滴加正丁基锂(2.6mmol),滴加完毕后,室温反应半小时,再向体系加入二芳基甲酮(2mmol),室温搅拌24h,TLC检测底物消失,反应结束。将饱和氯化铵水溶液(10mL)倒入反应中,用二氯甲烷(3×10mL)萃取,合并有机相,无水硫酸钠干燥、抽滤,然后减压蒸馏除去有机溶剂,经过硅胶柱层析(洗脱液为石油醚),得到无色油状液体,产物的结构经过NMR、MS证实为1b,收率为95%。
2)1,1-二芳基烷烃的制备
氮气保护下,向带有磁力搅拌装置的25mL圆底烧瓶中加入氯化亚铜(5mol%)、(S,S)-BDPP(5mol%)、NaBARF(5mol%)和叔丁醇钠(0.15mmol),再加入四氢呋喃(2ml),室温下搅拌10min。搅拌10min后,向体系中加入联硼酸频那醇酯(0.18mmol),继续搅拌10min,然后,加入1b(0.15mmol)和甲醇(0.3mmol)。将反应放在室温搅拌24h,并用TLC检测反应完成。反应用水(10ml)淬灭,二氯甲烷(3×10ml)萃取,合并有机相,用无水硫酸钠干燥、抽滤,加圧蒸留除去有机溶剂。最后经过硅胶柱层析,得到1,1-二芳基烷烃类化合物2b,产率为86%,ee值为95.5%。
图3为实施例2制备得到的1,1-二芳基硼化烷烃类化合物2b的1H-NMR的核磁共振谱;图4为实施例2制备得到的1,1-二芳基硼化烷烃类化合物2b的13C-NMR的核磁共振谱;图6为实施例2制备得到的1,1-二芳基硼化烷烃类化合物2b的高效液相色谱图。其中图a为反应配体为三苯基膦,且不加NarBARF,其它反应条件和实施例2相同,得到的1,1-二芳基烷烃类化合物的高效液相色谱图;图b为本发明实施例2制备得到的1,1-二芳基硼化烷烃类化合物2b的高效液相色谱图,具体数据如下:
1H NMR(500MHz,CDCl3)δ:7.36(d,J=8.0Hz,1H),7.22-7.16(m,5H),7.13-7.07(m,3H),4.46(t,J=8.5Hz,1H),2.29(s,3H),1.57(d,J=7.5Hz,2H),1.07(s,6H),1.02(s,6H);13C NMR(125MHz,CDCl3)δ:146.41,143.99,136.10,130.67,128.12,127.73,126.75,125.87,125.86,125.62,83.04,42.26,24.57,24.51,19.94.Mass Spectrometry:HRMS(ESI-TOF)(m/z):Calcd for C21H27BNaO2([M+Na]+):345.2000;found:345.1994.[α]D28=-20.6,(c=1.00,CHCl3).HPLC analysis(Chiralpak OD-H column,hexanes/i-PrOH=98/2,0.5ml/min,254nm,tR(minor)=8.4min,tR(major)=9.2min).
从图6可以看出,本发明制备的实施例2制备得到的1,1-二芳基硼化烷烃类化合物具有高对应选择性。
实施例3
1)1,1-二芳基乙烯1d的制备
氮气保护下,向带有磁力搅拌装置的25mL圆底烧瓶中加入甲基三苯基溴化膦(2.6mmol)和四氢呋喃(5ml)。将反应移到-78℃,向体系中滴加正丁基锂(2.6mmol),滴加完毕后,室温反应半小时,再向体系加入二芳基甲酮(2mmol),室温搅拌24h,TLC检测底物消失,反应结束。将饱和氯化铵水溶液(10mL)倒入反应中,用二氯甲烷(3×10mL)萃取,合并有机相,无水硫酸钠干燥、抽滤,然后减压蒸馏除去有机溶剂,经过硅胶柱层析(洗脱液为石油醚),得到无色油状液体,产物的结构经过NMR、MS证实为1d,收率为63%。
2)1,1-二芳基烷烃的制备
氮气保护下,向带有磁力搅拌装置的25mL圆底烧瓶中加入氯化亚铜(5mol%)、(S,S)-BDPP(5mol%)、NaBARF(5mol%)和叔丁醇钠(0.15mmol),再加入四氢呋喃(2ml),室温下搅拌10min。搅拌10min后,向体系中加入联硼酸频那醇酯(0.18mmol),继续搅拌10min,然后,加入1d(0.15mmol)和甲醇(0.3mmol)。将反应放在室温搅拌24h,并用TLC检测反应完成。反应用水(10ml)淬灭,二氯甲烷(3×10ml)萃取,合并有机相,用无水硫酸钠干燥、抽滤,加圧蒸留除去有机溶剂。最后经过硅胶柱层析,得到1,1-二芳基烷烃类化合物2d,产率为92%,ee值为93%。
谱图解析数据2d
1H NMR(500MHz,CDCl3)δ:7.37-7.35(m,1H),7.25-7.14(m,7H),7.12-7.09(m,1H),4.62(t,J=8.5Hz,1H),3.32-3.27(m,1H),1.58(d,J=8.5Hz,2H),1.23(d,J=7.0Hz,3H),1.06(s,6H),1.01(s,6H),0.95(d,J=7.0Hz,3H);13C NMR(125MHz,CDCl3)δ:147.23,146.69,142.13,128.09,127.63,127.11,126.28,125.52,125.43,125.35,83.03,41.48,28.17,24.55,24.51,24.20,23.63.Mass Spectrometry:HRMS(ESI-TOF)(m/z):Calcd for C23H31BNaO2([M+Na]+):373.2313;found:373.2302.[α]D27=-7.5,(c=1.33,CHCl3).HPLC analysis(Chiralpak OD-H column,hexanes/i-PrOH=98/2,0.5ml/min,254nm,tR(minor)=8.1min,tR(major)=9.4min).
实施例4
1)1,1-二芳基乙烯1e的制备
氮气保护下,向带有磁力搅拌装置的25mL圆底烧瓶中加入甲基三苯基溴化膦(2.6mmol)和四氢呋喃(5ml)。将反应移到-78℃,向体系中滴加正丁基锂(2.6mmol),滴加完毕后,室温反应半小时,再向体系加入二芳基甲酮(2mmol),室温搅拌24h,TLC检测底物消失,反应结束。将饱和氯化铵水溶液(10mL)倒入反应中,用二氯甲烷(3×10mL)萃取,合并有机相,无水硫酸钠干燥、抽滤,然后减压蒸馏除去有机溶剂,经过硅胶柱层析(洗脱液为石油醚),得到无色油状液体,产物的结构经过NMR、MS证实为1e,收率为36%。
2)1,1-二芳基烷烃的制备
氮气保护下,向带有磁力搅拌装置的25mL圆底烧瓶中加入氯化亚铜(5mol%)、(S,S)-BDPP(5mol%)、NaBARF(5mol%)和叔丁醇钠(0.15mmol),再加入四氢呋喃(2ml),室温下搅拌10min。搅拌10min后,向体系中加入联硼酸频那醇酯(0.18mmol),继续搅拌10min,然后,加入1e(0.15mmol)和甲醇(0.3mmol)。将反应放在室温搅拌24h,并用TLC检测反应完成。反应用水(10ml)淬灭,二氯甲烷(3×10ml)萃取,合并有机相,用无水硫酸钠干燥、抽滤,加圧蒸留除去有机溶剂。最后经过硅胶柱层析,得到1,1-二芳基烷烃类化合物2e,产率为45%,ee值为83%。
谱图解析数据2e
1H NMR(500MHz,CDCl3)δ:7.52(d,J=7.5Hz,1H),7.36-7.32(m,4H),7.22-7.19(m,3H),7.14-7.11(m,3H),7.07-7.04(m,1H),6.94(d,J=7.5Hz,2H),4.45-4.42(m,1H),1.65-1.60(m,1H),1.50-1.46(m,1H),1.03(s,6H),0.99(s,6H);13C NMR(125MHz,CDCl3)δ:147.18,143.01,141.87,130.04,129.52,127.96,127.76,127.46,127.38,127.16,126.65,125.70,125.42,82.98,41.98,29.70,24.59,24.44.Mass Spectrometry:HRMS(ESI-TOF)(m/z):Calcd for C26H29BNaO2([M+Na]+):407.2157;found:407.2165.[α]D28=+6.5,(c=0.39,CHCl3).HPLC analysis(Chiralpak OD-H column,hexanes/i-PrOH=98/2,0.5ml/min,254nm,tR(minor)=8.3min,tR(major)=9.4min).
实施例5
1)1,1-二芳基乙烯1g的制备
氮气保护下,向带有磁力搅拌装置的25mL圆底烧瓶中加入甲基三苯基溴化膦(2.6mmol)和四氢呋喃(5ml)。将反应移到-78℃,向体系中滴加正丁基锂(2.6mmol),滴加完毕后,室温反应半小时,再向体系加入二芳基甲酮(2mmol),室温搅拌24h,TLC检测底物消失,反应结束。将饱和氯化铵水溶液(10mL)倒入反应中,用二氯甲烷(3×10mL)萃取,合并有机相,无水硫酸钠干燥、抽滤,然后减压蒸馏除去有机溶剂,经过硅胶柱层析(洗脱液为V石油醚:V乙酸乙酯=50:1),得到无色油状液体,产物的结构经过NMR、MS证实为1g,收率为75%。
2)1,1-二芳基烷烃的制备
氮气保护下,向带有磁力搅拌装置的25mL圆底烧瓶中加入氯化亚铜(5mol%)、(S,S)-BDPP(5mol%)、NaBARF(5mol%)和叔丁醇钠(0.15mmol),再加入四氢呋喃(2ml),室温下搅拌10min。搅拌10min后,向体系中加入联硼酸频那醇酯(0.18mmol),继续搅拌10min,然后,加入1g(0.15mmol)和甲醇(0.3mmol)。将反应放在室温搅拌24h,并用TLC检测反应完成。反应用水(10ml)淬灭,二氯甲烷(3×10ml)萃取,合并有机相,用无水硫酸钠干燥、抽滤,加圧蒸留除去有机溶剂。最后经过硅胶柱层析,得到1,1-二芳基烷烃类化合物2g,产率为90%,ee值为88%。
谱图解析数据2g
1H NMR(500MHz,CDCl3)δ:7.39(d,J=7.0Hz,1H),7.31(d,J=7.5Hz,2H),7.22-7.07(m,6H),4.98(t,J=9.0Hz,1H),2.57(s,6H),1.58(d,J=9.0Hz,2H),1.02(s,6H),0.98(s,6H);13C NMR(125MHz,CDCl3)δ:152.74,147.52,141.52,128.23,127.96,127.65,126.61,125.33,123.95,120.49,82.87,45.69,39.17,24.50,24.45.Mass Spectrometry:HRMS(ESI-TOF)(m/z):Calcd for C22H31BNO2([M+H]+):352.2446;found:352.2462.[α]D28=-4.0,(c=0.50,CHCl3).HPLC analysis(Chiralpak AD-H column,hexanes/i-PrOH=98/2,0.5ml/min,254nm,tR(minor)=8.1min,tR(major)=10.3min).
实施例6
1)1,1-二芳基乙烯1i的制备
氮气保护下,向带有磁力搅拌装置的25mL圆底烧瓶中加入甲基三苯基溴化膦(2.6mmol)和四氢呋喃(5ml)。将反应移到-78℃,向体系中滴加正丁基锂(2.6mmol),滴加完毕后,室温反应半小时,再向体系加入二芳基甲酮(2mmol),室温搅拌24h,TLC检测底物消失,反应结束。将饱和氯化铵水溶液(10mL)倒入反应中,用二氯甲烷(3×10mL)萃取,合并有机相,无水硫酸钠干燥、抽滤,然后减压蒸馏除去有机溶剂,经过硅胶柱层析(洗脱液为石油醚),得到白色固体,产物的结构经过NMR、MS证实为1i,收率为68%。
2)1,1-二芳基烷烃的制备
氮气保护下,向带有磁力搅拌装置的25mL圆底烧瓶中加入氯化亚铜(5mol%)、(S,S)-BDPP(5mol%)、NaBARF(5mol%)和叔丁醇钠(0.15mmol),再加入四氢呋喃(2ml),室温下搅拌10min。搅拌10min后,向体系中加入联硼酸频那醇酯(0.18mmol),继续搅拌10min,然后,加入1i(0.15mmol)和甲醇(0.3mmol)。将反应放在室温搅拌24h,并用TLC检测反应完成。反应用水(10ml)淬灭,二氯甲烷(3×10ml)萃取,合并有机相,用无水硫酸钠干燥、抽滤,加圧蒸留除去有机溶剂。最后经过硅胶柱层析,得到1,1-二芳基烷烃类化合物2i,产率为98%,ee值为88%。
谱图解析数据2i
1H NMR(500MHz,CDCl3)δ:8.16(d,J=9.0Hz,1H),7.82-7.80(m,1H),7.70(d,J=8.0Hz,1H),7.52(d,J=7.0Hz,1H),7.46-7.40(m,3H),7.29(d,J=7.0Hz,2H),7.21(t,J=8.0Hz,2H),7.12-7.09(m,1H),5.09(t,J=8.0Hz,1H),1.76-1.66(m,2H),1.06(s,6H),0.98(s,6H);13C NMR(125MHz,CDCl3)δ:146.82,141.87,133.89,131.69,128.56,128.23,127.68,126.80,125.79,125.65,125.29,125.18,124.31,124.25,83.11,41.89,24.53.Mass Spectrometry:HRMS(ESI-TOF)(m/z):Calcd for C24H27BNaO2([M+Na]+):381.2001;found:381.1999.[α]D27=+2.9,(c=0.52,CHCl3).HPLC analysis(Chiralpak OD-H column,hexanes/i-PrOH=98/2,0.5ml/min,254nm,tR(minor)=11.8min,tR(major)=13.0min).
实施例7
1)1,1-二芳基乙烯1k的制备
氮气保护下,向带有磁力搅拌装置的25mL圆底烧瓶中加入甲基三苯基溴化膦(2.6mmol)和四氢呋喃(5ml)。将反应移到-78℃,向体系中滴加正丁基锂(2.6mmol),滴加完毕后,室温反应半小时,再向体系加入二芳基甲酮(2mmol),室温搅拌24h,TLC检测底物消失,反应结束。将饱和氯化铵水溶液(10mL)倒入反应中,用二氯甲烷(3×10mL)萃取,合并有机相,无水硫酸钠干燥、抽滤,然后减压蒸馏除去有机溶剂,经过硅胶柱层析(洗脱液为石油醚),得到无色油状液体,产物的结构经过NMR、MS证实为1k,收率为57%。
2)1,1-二芳基烷烃的制备
氮气保护下,向带有磁力搅拌装置的25mL圆底烧瓶中加入氯化亚铜(5mol%)、(S,S)-BDPP(5mol%)、NaBARF(5mol%)和叔丁醇钠(0.15mmol),再加入四氢呋喃(2ml),室温下搅拌10min。搅拌10min后,向体系中加入联硼酸频那醇酯(0.18mmol),继续搅拌10min,然后,加入1k(0.15mmol)和甲醇(0.3mmol)。将反应放在室温搅拌24h,并用TLC检测反应完成。反应用水(10ml)淬灭,二氯甲烷(3×10ml)萃取,合并有机相,用无水硫酸钠干燥、抽滤,加圧蒸留除去有机溶剂。最后经过硅胶柱层析,得到1,1-二芳基烷烃类化合物2k,产率为78%,ee值为87%。
谱图解析数据2k
1H NMR(500MHz,CDCl3)δ:7.24(d,J=7.5Hz,1H),7.17-7.05(m,4H),6.91-6.90(m,1H),6.87(d,J=5.0Hz 1H),4,52(t,J=8.5Hz,1H),2.36(s,3H),1.62-1.51(m,2H),1.06(s,6H),1.04(s,6H);13C NMR(125MHz,CDCl3)δ:147.56,144.12,135.64,130.13,127.80,126.77,126.05,125.90,125.04,119.82,83.06,37.83,24.55,24.54,19.74.Mass Spectrometry:HRMS(ESI-TOF)(m/z):Calcd for C19H25BNaO2S([M+Na]+):351.1564;found:351.1574.[α]D28=+16.9,(c=0.33,CHCl3).HPLC analysis(Chiralpak AD-H column,hexanes/i-PrOH=98/2,0.5ml/min,254nm,tR(minor)=8.7min,tR(major)=11.7min).
实施例8
1)1,1-二芳基乙烯1l的制备
氮气保护下,向带有磁力搅拌装置的25mL圆底烧瓶中加入甲基三苯基溴化膦(2.6mmol)和四氢呋喃(5ml)。将反应移到-78℃,向体系中滴加正丁基锂(2.6mmol),滴加完毕后,室温反应半小时,再向体系加入二芳基甲酮(2mmol),室温搅拌24h,TLC检测底物消失,反应结束。将饱和氯化铵水溶液(10mL)倒入反应中,用二氯甲烷(3×10mL)萃取,合并有机相,无水硫酸钠干燥、抽滤,然后减压蒸馏除去有机溶剂,经过硅胶柱层析(洗脱液为V石油醚:V乙醚=20:1),得到无色油状液体,产物的结构经过NMR、MS证实为1l,收率为40%。
2)1,1-二芳基烷烃的制备
氮气保护下,向带有磁力搅拌装置的25mL圆底烧瓶中加入氯化亚铜(5mol%)、(S,S)-BDPP(5mol%)、NaBARF(5mol%)和叔丁醇钠(0.15mmol),再加入四氢呋喃(2ml),室温下搅拌10min。搅拌10min后,向体系中加入联硼酸频那醇酯(0.18mmol),继续搅拌10min,然后,加入1l(0.15mmol)和甲醇(0.3mmol)。将反应放在室温搅拌24h,并用TLC检测反应完成。反应用水(10ml)淬灭,二氯甲烷(3×10ml)萃取,合并有机相,用无水硫酸钠干燥、抽滤,加圧蒸留除去有机溶剂。最后经过硅胶柱层析,得到1,1-二芳基烷烃类化合物2l,产率为31%,ee值为91%。
谱图解析数据2l
1H NMR(500MHz,CDCl3)δ:7.44-7.39(m,2H),7.19-7.15(m,2H),7.08-7.05(m,3H),6.96(d,J=3.0Hz,1H),6.36(d,J=3.0Hz,1H),4.58(t,J=8.5Hz,1H),3.71(s,3H),2.30(s,3H),1.63-1.61(m,2H),1.07(s,6H),1.01(s,6H);13C NMR(125MHz,CDCl3)δ:144.92,137.54,136.16,135.18,130.08,128.65,128.30,126.88,125.73,125.59,122.07,119.36,108.82,100.67,82.95,42.29,32.77,24.58,24.55,19.97.Mass Spectrometry:HRMS(ESI-TOF)(m/z):Calcd for C24H30BNNaO2([M+Na]+):398.2266;found:398.2261.[α]D28=-26.7,(c=0.33,CHCl3).HPLC analysis(Chiralpak OD-H column,hexanes/i-PrOH=98/2,0.5ml/min,254nm,tR(minor)=19.1min,tR(major)=15.0min).
实施例9
1)1,1-二芳基乙烯1p的制备
氮气保护下,向带有磁力搅拌装置的25mL圆底烧瓶中加入甲基三苯基溴化膦(2.6mmol)和四氢呋喃(5ml)。将反应移到-78℃,向体系中滴加正丁基锂(2.6mmol),滴加完毕后,室温反应半小时,再向体系加入二芳基甲酮(2mmol),室温搅拌24h,TLC检测底物消失,反应结束。将饱和氯化铵水溶液(10mL)倒入反应中,用二氯甲烷(3×10mL)萃取,合并有机相,无水硫酸钠干燥、抽滤,然后减压蒸馏除去有机溶剂,经过硅胶柱层析(洗脱液为V石油醚:V乙醚=20:1),得到无色油状液体,产物的结构经过NMR、MS证实为1p,收率为56%。
2)1,1-二芳基烷烃的制备
氮气保护下,向带有磁力搅拌装置的25mL圆底烧瓶中加入氯化亚铜(5mol%)、(S,S)-BDPP(5mol%)、NaBARF(5mol%)和叔丁醇钠(0.15mmol),再加入四氢呋喃(2ml),室温下搅拌10min。搅拌10min后,向体系中加入联硼酸频那醇酯(0.18mmol),继续搅拌10min,然后,加入1p(0.15mmol)和甲醇(0.3mmol)。将反应放在室温搅拌24h,并用TLC检测反应完成。反应用水(10ml)淬灭,二氯甲烷(3×10ml)萃取,合并有机相,用无水硫酸钠干燥、抽滤,加圧蒸留除去有机溶剂。最后经过硅胶柱层析,得到1,1-二芳基烷烃类化合物2p,产率为84%,ee值为84%。
谱图解析数据2p
1H NMR(500MHz,CDCl3)δ:7.17(d,J=8.0Hz,1H),7.06(t,J=7.5Hz,1H),6.98(d,J=7.5Hz,1H),6.78(s,1H),6.73(s,2H),4.48(t,J=8.5Hz,1H),3.82(s,3H),3.81(s,3H),2.25(s,3H),2.21(s,3H),1.53(dd,J=8.5,2.0Hz,2H),1.07(s,6H),1.04(s,6H);13C NMR(125MHz,CDCl3)δ:148.51,146.90,144.22,139.47,136.50,134.43,127.64,125.19,124.60,119.68,111.40,110.88,83.02,55.83,55.72,42.15,24.61,24.56,21.01,15.16.Mass Spectrometry:HRMS(ESI-TOF)(m/z):Calcd for C24H33BNaO4([M+Na]+):419.2368;found:419.2352.[α]D28=+7.6,(c=0.68,CHCl3).HPLC analysis(Chiralpak AD-H column,hexanes/i-PrOH=95/5,0.5ml/min,254nm,tR(minor)=15.1min,tR(major)=11.4min);
实施例10
1)1,1-二芳基烯烃1q的制备
向带有磁力搅拌装置的25mL圆底烧瓶中加入底物(2mmol)、对甲苯磺酸(6.0mmol)和四氢呋喃(5ml),加热到120℃,回流6h,TLC检测底物消失,反应结束。将饱和氯化铵水溶液(10mL)倒入反应中,用二氯甲烷(3×10mL)萃取,合并有机相,无水硫酸钠干燥、抽滤,然后减压蒸馏除去有机溶剂,经过硅胶柱层析(洗脱液为石油醚),得到无色油状液体,产物的结构经过NMR、MS证实为1q,收率为45%。
2)1,1-二芳基烷烃的制备
氮气保护下,向带有磁力搅拌装置的25mL圆底烧瓶中加入氯化亚铜(5mol%)、(S,S)-BDPP(5mol%)、NaBARF(5mol%)和叔丁醇钠(0.15mmol),再加入四氢呋喃(2ml),室温下搅拌10min。搅拌10min后,向体系中加入联硼酸频那醇酯(0.18mmol),继续搅拌10min,然后,加入1q(0.15mmol)和甲醇(0.3mmol)。将反应放在室温搅拌24h,并用TLC检测反应完成。反应用水(10ml)淬灭,二氯甲烷(3×10ml)萃取,合并有机相,用无水硫酸钠干燥、抽滤,加圧蒸留除去有机溶剂。最后经过硅胶柱层析,得到1,1-二芳基烷烃类化合物2q,产率为73%,ee值为89%。
谱图解析数据2q
1H NMR(500MHz,CDCl3)δ:7.30-7.09(m,8H),6.90(d,J=7.5Hz,1H),4.44(d,J=11.0Hz,1H),3.18-3.13(m,1H),3.01-2.96(m,1H),1.90-1.83(m,1H),1.23(s,6H),1.22(s,6H);13C NMR(125MHz,CDCl3)δ:147.35,145.42,144.64,128.41,128.23,126.27,126.16,126.13,124.75,123.98,83.26,54.33,34.58,24.84,24.65.Mass Spectrometry:HRMS(ESI-TOF)(m/z):Calcd for C21H25BNaO2([M+Na]+):343.1844;found:343.1840.[α]D28=+10.0,(c=0.40,CHCl3).HPLC analysis(Chiralpak OD-H column,hexanes/i-PrOH=98/2,0.5ml/min,254nm,tR(minor)=8.0min,tR(major)=7.3min).
实施例11
1)烯烃1r的制备
氮气保护下,向带有磁力搅拌装置的25mL圆底烧瓶中加入甲基三苯基溴化膦(2.6mmol)和四氢呋喃(5ml)。将反应移到-78℃,向体系中滴加正丁基锂(2.6mmol),滴加完毕后,室温反应半小时,再向体系加入2-萘乙酮(2mmol),室温搅拌24h,TLC检测底物消失,反应结束。将饱和氯化铵水溶液(10mL)倒入反应中,用二氯甲烷(3×10mL)萃取,合并有机相,无水硫酸钠干燥、抽滤,然后减压蒸馏除去有机溶剂,经过硅胶柱层析(洗脱液为石油醚),得到无色油状液体,产物的结构经过NMR、MS证实为1r,收率为84%。
2)芳硼烷2r的制备
氮气保护下,向带有磁力搅拌装置的25mL圆底烧瓶中加入氯化亚铜(5mol%)、(S,S)-BDPP(5mol%)、NaBARF(5mol%)和叔丁醇钠(0.15mmol),再加入四氢呋喃(2ml),室温下搅拌10min。搅拌10min后,向体系中加入联硼酸频那醇酯(0.18mmol),继续搅拌10min,然后,加入1r(0.15mmol)和甲醇(0.3mmol)。将反应放在室温搅拌24h,并用TLC检测反应完成。反应用水(10ml)淬灭,二氯甲烷(3×10ml)萃取,合并有机相,用无水硫酸钠干燥、抽滤,加圧蒸留除去有机溶剂。最后经过硅胶柱层析,得到1,1-二芳基烷烃类化合物2r,产率为77%,ee值为86%。
谱图解析数据2r
1H NMR(500MHz,CDCl3)δ:7.78-7.75(m,3H),7.65(s,1H),7.44-7.37(m,3H),3.23-3.19(m,1H),1.36(d,J=7.0Hz,3H),1.30-1.20(m,2H),1.14(s,12H);13C NMR(125MHz,CDCl3)δ:146.67,133.55,132.05,127.71,127.54,127.49,125.81,125.64,124.89,124.36,83.00,35.85,24.74,24.69.Mass Spectrometry:HRMS(ESI-TOF)(m/z):Calcd for C19H25BNaO2([M+Na]+):319.1843;found:319.1833.[α]D28=-25.0,(c=0.48,CHCl3).HPLC analysis(Chiralpak OJ-H column,hexanes/i-PrOH=99/1,0.5ml/min,254nm,tR(minor)=14.6min,tR(major)=15.1min).
实施例12
1)1,1-二芳基乙烯1s的制备
氮气保护下,向带有磁力搅拌装置的25mL圆底烧瓶中加入甲基三苯基溴化膦(2.6mmol)和四氢呋喃(5ml)。将反应移到-78℃,向体系中滴加正丁基锂(2.6mmol),滴加完毕后,室温反应半小时,再向体系加入二芳基甲酮(2mmol),室温搅拌24h,TLC检测底物消失,反应结束。将饱和氯化铵水溶液(10mL)倒入反应中,用二氯甲烷(3×10mL)萃取,合并有机相,无水硫酸钠干燥、抽滤,然后减压蒸馏除去有机溶剂,经过硅胶柱层析(洗脱液为V石油醚:V乙醚=20:1),得到无色油状液体,产物的结构经过NMR、MS证实为1s,收率为89%。
2)1,1-二芳基烷烃的制备
氮气保护下,向带有磁力搅拌装置的25mL圆底烧瓶中加入氯化亚铜(5mol%)、(S,S)-BDPP(5mol%)、NaBARF(5mol%)和叔丁醇钠(0.15mmol),再加入四氢呋喃(2ml),室温下搅拌10min。搅拌10min后,向体系中加入联硼酸频那醇酯(0.18mmol),继续搅拌10min,然后,加入1s(0.15mmol)和甲醇(0.3mmol)。将反应放在室温搅拌24h,并用TLC检测反应完成。反应用水(10ml)淬灭,二氯甲烷(3×10ml)萃取,合并有机相,用无水硫酸钠干燥、抽滤,加圧蒸留除去有机溶剂。最后经过硅胶柱层析,得到1,1-二芳基烷烃类化合物2s,产率为86%,ee值为92%。
谱图解析数据2s
1H NMR(600MHz,CDCl3)δ:7.10-7.02(m,4H),6.93-6.91(m,1H),6.78(s,1H),6.63(d,J=8.4Hz,1H),6.47(d,J=8.4Hz,1H),4.52(t,J=7.8Hz,1H),2.18(s,3H),1.46-1.58(m,2H),1.07(s,6H),1.02(s,6H),0.92(s,9H),0.25(s,3H),0.19(s,3H);13C NMR(125 MHz,CDCl3)δ:150.62,146.64,136.76,129.77,128.88,127.87,127.83,126.85,125.40,117.94,82.82,38.85,25.96,24.54,20.75,18.34,-4.01,-4.08.Mass Spectrometry:HRMS(ESI-TOF)(m/z):Calcd for C27H41BNaO3Si([M+Na]+):475.2815;found:475.2824.HPLC analysis(Chiralpak AD-H column,hexanes/i-PrOH=98/2,0.5 ml/min,254 nm,tR(minor)=4.1 min,tR(major)=18.7min).
- 还没有人留言评论。精彩留言会获得点赞!