新化合物及其制造方法、以及其用途与流程
技术领域
本发明涉及新化合物、其制造方法、以及含有所述新化合物的组合物、抗癌剂、和抗幽门螺杆菌(Helicobacter pylori)剂。
背景技术:
癌组织以癌细胞和被称为间质的周围正常组织混在一起的形式构成。以下情况逐渐明确:所述间质由血管、细胞外基质、成纤维细胞样细胞(也有时简称为“间质细胞”)等各种各样的因子构成,并与癌的增殖密切相关。已知所述间质之中、特别是间质细胞介由粘接、分泌因子以正、负来控制癌细胞的增殖(例如参照非专利文献1)。这种状况下,进行更有用的新抗癌剂的探索,并强烈要求其的快速提供。
在胃溃疡、十二指肠溃疡等胃和十二指肠损害中,已知有由幽门螺杆菌(Helicobacter pylori)引发的。于是,提出了喹啉酮化合物作为具有抗幽门螺杆菌活性的化合物(例如参照非专利文献2和专利文献1)。然而,不能说所述提出的喹啉酮化合物用作药品是足够的,要求新的具有抗幽门螺杆菌活性的化合物。
现有技术文献
专利文献
专利文献1:美国专利第5942619号说明书
非专利文献
非专利文献1:Kawada,M.,Inoue,H.,Masuda,T.,and Ikeda,D.Insulin-like growth factor-I secreted from prostate stromal cells mediates tumor-stromal cell interactions of the prostate cancer.Cancer Res.66,4419-4425(2006).
非专利文献2:Dekker,K.A.,Inagaki,T.,Gootz,T.D.,Huang,L.H.,Kojima Y.,Kohlbrenner,W.E.,Matsunaga,Y.,McGuirk,P.R.,Nomura,E.,Sakakibara,T.Sakemi S.,Suzuki,Y.,Yamauchi,Y.,and Kojima,N.New quinolone compounds from Pseudonocardia sp.with selective and potent anti-Helicobacter pylori activity:taxonomy of producing strain,fermentation,isolation,structural elucidation and biological activities.J.Antibiot.51,145-152(1998).
技术实现要素:
发明所要解决的课题
本发明是鉴于上述以往技术而进行的,其课题在于实现以下目的。即,本发明的目的在于,提供具有优异的抗癌作用、或优异的抗幽门螺杆菌活性的新化合物、和该新化合物的制造方法、以及利用所述新化合物的含化合物的组合物、抗癌剂、和抗幽门螺杆菌剂。
解决技术问题的手段
作为解决所述课题的手段,如下。即,
<1>一种化合物,其特征在于,以下述结构式(1)~(13)中的任一个表示。
其中,所述结构式(1)~(13)中,Me表示甲基。
<2>下述结构式(3)、(4)、和(8)中的任一个表示的化合物的制造方法,其特征在于,使下述结构式(17)、(18)、和(19)中的任一个表示的化合物、与碱金属的氢氧化物和碱土类金属的氢氧化物中的至少一个进行反应。
<3>下述结构式(5)表示的化合物的制造方法,其特征在于,使下述结构式(21)表示的化合物、与碱金属的醇盐、碱金属的碳酸盐和碱金属的氢化物中的至少一个反应后,使所述反应物与溴乙酸甲酯进行反应。
其中,所述结构式(5)中,Me表示甲基。
<4>下述结构式(6)表示的化合物的制造方法,其特征在于,使下述结构式(5)表示的化合物、与碱金属的氢氧化物和碱土类金属的氢氧化物中的至少一个反应后,使所述反应物的pH为酸性。
其中,所述结构式(5)中,Me表示甲基。
<5>下述结构式(1)表示的化合物的制造方法,其特征在于,使下述结构式(21)表示的化合物、与碱金属的碳酸盐和碱金属的氢化物中的至少一个、与溴乙酸甲酯进行反应。
其中,所述结构式(1)中,Me表示甲基。
<6>下述结构式(2)表示的化合物的制造方法,其特征在于,使下述结构式(1)表示的化合物、与碱金属的氢氧化物和碱土类金属的氢氧化物中的至少一个反应后,使所述反应物的pH为酸性。
其中,所述结构式(1)中,Me表示甲基。
<7>下述结构式(7)表示的化合物的制造方法,其特征在于,使下述结构式(21)表示的化合物、与碱金属的醇盐和碱金属的氢化物中的至少一个反应后,使所述反应物与溴化氰进行反应。
<8>下述结构式(9)表示的化合物的制造方法,其特征在于,使下述结构式(6)表示的化合物、与叔胺和吡啶类中的至少一个、与叠氮磷酸二苯酯、与甲硫醇钠进行反应。
其中,所述结构式(9)中,Me表示甲基。
<9>下述结构式(10)表示的化合物的制造方法,其特征在于,使下述结构式(6)表示的化合物、与叔胺和吡啶类中的至少一个、与叠氮磷酸二苯酯反应后,使所述反应物与甲硫醇钠进行反应。
其中,所述结构式(10)中,Me表示甲基。
<10>下述结构式(11)表示的化合物的制造方法,其特征在于,使下述结构式(20)表示的化合物、与碱金属的醇盐和碱金属的氢化物中的至少一个反应后,使所述反应物与硫氰酸氯甲酯进行反应。
<11>下述结构式(12)表示的化合物的制造方法,其特征在于,在乙腈的存在下,使下述结构式(11)表示的化合物、与甲硫醇钠进行反应。
其中,所述结构式(12)中,Me表示甲基。
<12>下述结构式(13)表示的化合物的制造方法,其特征在于,在乙腈的存在下,使下述结构式(11)表示的化合物、与甲硫醇钠反应后,使所述反应物与甲基化试剂进行反应。
其中,所述结构式(13)中,Me表示甲基。
<13>一种含化合物的组合物,其特征在于,含有所述<1>所述的化合物。
<14>一种抗癌剂,其特征在于,含有所述<1>所述的化合物。
<15>一种抗幽门螺杆菌剂,其特征在于,含有所述<1>所述的化合物。
<16>一种用于预防或治疗癌的方法,其特征在于,对个体给予所述<14>所述的抗癌剂。
<17>一种用于预防或治疗由幽门螺杆菌带来的感染病的方法,其特征在于,对个体给予所述<15>所述的抗幽门螺杆菌剂。
<18>一种用于预防或治疗由幽门螺杆菌引起的胃和十二指肠损害的方法,其特征在于,对个体给予所述<15>所述的抗幽门螺杆菌剂。
发明效果
根据本发明,能够实现所述目的,能够提供具有优异的抗癌作用、或优异的抗幽门螺杆菌活性的新化合物、和该新化合物的制造方法、以及利用所述新化合物的含化合物的组合物、抗癌剂、和抗幽门螺杆菌剂。
附图说明
图1A:图1A是表示试验例2-1中的肿瘤体积的变化的图表。
图1B:图1B是表示试验例2-1中的肿瘤重量(从肿瘤接种起第21天)的图表。
图1C:图1C是表示试验例2-2中的肿瘤体积的变化的图表。
图1D:图1D是表示试验例2-2中的肿瘤重量(从肿瘤接种起第21天)的图表。
具体实施方式
(新化合物)
本发明的化合物是下述结构式(1)~(13)表示的化合物,是本发明者们发现的新化合物。
其中,所述结构式(1)~(13)中,Me表示甲基。
<结构式(1)表示的化合物的理化性质>
作为所述结构式(1)表示的化合物的理化性质,如下。
(1)外观:无色油状物
(2)分子式:C21H29O3N
(3)高分辨质谱分析(HRESI-MS)(m/z):
实验值344.2221(M+H)+
计算值344.2220(以C21H30O3N计)
(4)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:2953,2925,2854,1765,1618,1596,1437,1123,968,768,680
(5)质子核磁共振光谱(400MHz,CDCl3):
δ=0.87(3H,t,J=6.6),1.20-1.48(10H,m),1.71(2H,m),2.43(3H,s),2.95(2H,m),3.86(3H,s),4.62(2H,s),7.47(1H,ddd,J=8.2,6.8,1.1),7.62(1H,ddd,J=8.4,6.8,1.3),8.00(1H,d,J=8.4),8.05(1H,d,J=8.2)
(6)碳13核磁共振光谱(100MHz,CDCl3):
δ=11.90,14.08,22.63,28.99,29.23,29.49,29.86,31.83,37.02,52.32,70.26,120.85,120.73,121.39,121.76,125.73,128.82,128.85,147.84,159.03,164.32,168.95
<结构式(2)表示的化合物的理化性质>
作为所述结构式(2)表示的化合物的理化性质,如下。
(1)外观:白色粉状
(2)熔点:59℃~62℃
(3)分子式:C20H27O3N
(4)高分辨质谱分析(HRESI-MS)(m/z):
实验值330.2064(M+H)+
计算值330.2064(以C20H28O3N计)
(5)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:2927,2855,2713,1736,1642,1589,1227,1181,1078,764,724
(6)质子核磁共振光谱(600MHz,Methanol-d4):
δ=0.88(3H,t,J=6.8),1.25-1.41(8H,m),1.50(2H,m),1.78(2H,m),2.54(3H,s),3.15(2H,t,m),4.92(2H,s),7.54(1H,ddd,J=8.2,7.2,1.0),7.92(1H,ddd,J=8.5,6.8,1.0),8.07(1H,brd,J=8.5),8.42(1H,brd,J=8.2)
(7)碳13核磁共振光谱(150MHz,Methanol-d4):
δ=12.23,14.40,23.67,29.93,30.27,30.34,30.74,32.97,35.07,72.72,122.81,123.71,123.85,124.62,129.16,133.85,142.14,164.30,167.46,171.91
<结构式(3)表示的化合物的理化性质>
作为所述结构式(3)表示的化合物的理化性质,如下。
(1)外观:白色粉状
(2)熔点:178℃~181℃
(3)分子式:C19H25ON
(4)高分辨质谱分析(HRESI-MS)(m/z):
实验值284.2011(M+H)+
计算值284.2009(以C19H26ON计)
(5)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:3064,2957,2933,1670,1638,1614,1555,1500,1371,1358,1152,1028,998,967,756,691
(6)质子核磁共振光谱(600MHz,CDCl3):
δ=0.95(6H,d,J=6.5),1.44(2H,m),1.69(2H,m),2.01(2H,q,J=6.8),2.15(3H,s),2.21(1H,m),2.70(2H,m),5.27-5.32(1H,m),5.36-5.39(1H,m),7.28(1H,ddd,J=8.2,5.8,1.0),7.32(1H,brd,J=8.2),7.52(1H,ddd,J=8.2,5.5,1.4),8.36(1H,dd,J=8.2,1.4),8.65(1H,br)
(7)碳13核磁共振光谱(150MHz,CDCl3):
δ=10.65,22.64,27.77,29.22,30.98,32.09,32.96,115.72,116.69,123.00,123.66,126.14,126.30,131.25,138.49,138.78,148.54,178.16
<结构式(4)表示的化合物的理化性质>
作为所述结构式(4)表示的化合物的理化性质,如下。
(1)外观:白色粉状
(2)熔点:240℃~244℃;
(3)分子式:C14H17ON
(4)高分辨质谱分析(HRESI-MS)(m/z):
实验值216.1385(M+H)+
计算值216.1383(以C14H18ON计)
(5)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:3059,2956,1636,1609,1554,1505,1369,1359,1189,998,762,695
(6)质子核磁共振光谱(400MHz,DMSO-d6):
δ=0.89(6H,d,J=6.6),1.94(3H,s),1.99(1H,m),2.53(2H,d,J=7.5),7.18(1H,ddd,J=8.2,6.6,1.4),7.46(1H,d,J=8.2),7.52(1H,ddd,J=8.2,6.6,1.4),8.01(1H,dd,J=8.2,1.1),11.23(1H,brs)
(7)碳13核磁共振光谱(100MHz,DMSO-d6):
δ=11.03,22.28,28.30,114.68,117.74,122.39,123.00,125.18,131.08,139.33,148.76,176.43
<结构式(5)表示的化合物的理化性质>
作为所述结构式(5)表示的化合物的理化性质,如下。
(1)外观:无色油状物
(2)分子式:C21H29O3N
(3)高分辨质谱分析(HRESI-MS)(m/z):
实验值344.2222(M+H)+
计算值344.2220(以C21H30O3N计)
(4)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:2953,2922,1743,1635,1617,1558,1507,1214,994,760,688
(5)质子核磁共振光谱(400MHz,CDCl3):
δ=0.89(3H,t,J=6.6),1.21-1.51(10H,m),1.60(2H,m),2.22(3H,s),2.74(2H,br),3.80(3H,s),4.90(2H,s),7.20(1H,d,J=8.7),7.33(1H,ddd,J=8.0,6.8,0.7),7.58(1H,ddd,J=8.7,
(6)碳13核磁共振光谱(100MHz,CDCl3):
δ=11.65,14.06,22.60,28.06,29.13,29.17,29.75,30.93,31.75,48.47,53.01,114.19,117.55,123.11,124.79,127.30,131.89,140.66,150.66,168.69,177.39
<结构式(6)表示的化合物的理化性质>
作为所述结构式(6)表示的化合物的理化性质,如下。
(1)外观:白色粉状
(2)熔点:161℃~163℃
(3)分子式:C20H27O3N
(4)高分辨质谱分析(HRESI-MS)(m/z):
实验值330.2063(M+H)+
计算值330.2064(以C20H28O3N计)
(5)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:2955,2925,2853,1725,1635,1593,1506,1191,976,760,689
(6)质子核磁共振光谱(400MHz,CDCl3):
δ=0.87(3H,t,J=6.4),1.20-1.65(12H,m),2.19(3H,s),2.80(2H,br),4.98(2H,br),7.26(1H,t,J=8.0),7.42(1H,d,J=8.7),7.54(1H,ddd,J=8.7,6.8,1.1),8.39(1H,dd,J=8.0,1.1)
(7)碳13核磁共振光谱(100MHz,CDCl3):
δ=11.91,14.06,22.68,27.85,29.12,29.78,31.26,31.73,49.71,115.46,117.21,123.86,123.94,126.69,132.47,140.53,154.22,169.35,176.57
<结构式(7)表示的化合物的理化性质>
作为所述结构式(7)表示的化合物的理化性质,如下。
(1)外观:无色油状物
(2)分子式:C19H24ON2
(3)高分辨质谱分析(HRESI-MS)(m/z):
实验值297.1961(M+H)+
计算值297.1961(以C19H25ON2计)
(4)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:2961,2926,2853,2237,1628,1576,1470,1292,1191,761,693
(5)质子核磁共振光谱(400MHz,CDCl3):
δ=0.87(3H,t,J=6.8),1.20-1.50(10H,m),1.71(2H,m),2.15(3H,s),2.93(2H,m),7.47(1H,m),7.73(2H,m),8.33(1H,ddd,J=8.0,0.92,1.1)
(6)碳13核磁共振光谱(100MHz,CDCl3):
δ=11.20,14.05,22.59,28.00,29.07,29.11,29.42,31.73,31.80,106.42,116.28,120.30,123.35,126.23,127.08,133.34,137.25,146.10.177.31
<结构式(8)表示的化合物的理化性质>
作为所述结构式(8)表示的化合物的理化性质,如下。
(1)外观:黄色粉状
(2)熔点:>260℃
(3)分子式:C18H15ON
(4)高分辨质谱分析(HRESI-MS)(m/z):
实验值284.1046(M+Na)+
计算值284.1046(以C18H15ONNa计)
(5)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:3064,2938,1628,1570,1507,1387,1359,1187,965,755,690
(6)质子核磁共振光谱(400MHz,DMSO-d6):
δ=2.13(3H,s),7.21(1H,ddd,J=8.5,6.8,1.1),7.32-7.50(5H,m),7.55-7.59(1H,m),7.67-7.72(3H,m),8.03(1H,dd,J=8.2,1.4),11.20(1H,s)
(7)碳13核磁共振光谱(100MHz,DMSO-d6):
δ=10.66,115.68,118.18,121.14,122.55,123.13,125.14,127.57,129.16,129.30,131.61,135.11,135.94,139.75,143.14,176.76
<结构式(9)表示的化合物的理化性质>
作为所述结构式(9)表示的化合物的理化性质,如下。
(1)外观:无色油状物
(2)分子式:C21H29O2NS
(3)高分辨质谱分析(HRESI-MS)(m/z):
实验值360.1994(M+H)+
计算值360.1992(以C21H30O2NS计)
(4)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:2924,2852,1687,1614,1594,1542,1193,1028,757,558
(5)质子核磁共振光谱(400MHz,CDCl3):
δ=0.88(3H,t,J=6.4),1.20-1.50(10H,m),1.60(2H,br),2.23(3H,s),2.33(3H,s),2.51-2.99(2H,br),5.01(2H,br),7.21(1H,d,J=8.7),7.34(1H,dd,J=8.0、6.6),7.58(1H,ddd,J=8.7,6.6,1.4),8.47(1H,dd,J=8.0,1.4)
(6)碳13核磁共振光谱(100MHz,CDCl3):
δ=11.38,11.68,14.06,22.60,28.16,29.13,29.16,29.75,31.03,31.74,55.95,114.61,117.94,123.32,127.29,131.97,132.02,140.73,150.65,177.49,196.82
<结构式(10)表示的化合物的理化性质>
作为所述结构式(10)表示的化合物的理化性质,如下。
(1)外观:无色油状物
(2)分子式:C21H30O2N2S
(3)高分辨质谱分析(HRESI-MS)(m/z):
实验值397.1921(M+Na)+
计算值397.1920(以C21H30O2N2NaS计)
(4)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:3169,2955,2927,1671,1615,1595,1556,1492,1195,1084,760,651
(5)质子核磁共振光谱(600MHz,CDCl3):
δ=0.89(3H,t,J=6.7),1.22-1.45(15H,m),2.45(2H,br),2.46(3H,s),5.67(2H,br),7.24(1H,ddd,J=7.9,6.8,1.0),7.49(1H,d,J=8.6),7.59(1H,ddd,J=8.6,6.8,1.4),8.26(1H,dd,J=7.9,1.4),8.78(1H,br)
(6)碳13核磁共振光谱(150MHz,CDCl3):
δ=11.05,12.22,14.02,22.59,28.48,29.11,29.16,29.73,30.73,31.77,52.58,115.46,117.00,123.24,124.37,126.89,132.32,139.56,151.60,168.61,177.27
<结构式(11)表示的化合物的理化性质>
作为所述结构式(11)表示的化合物的理化性质,如下。
(1)外观:黄色油状物
(2)分子式:C19H24ON2S
(3)高分辨质谱分析(HRESI-MS)(m/z):
实验值329.1682(M+H)+
计算值329.1682(以C19H25ON2S计)
(4)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:2961,2926,2853,2237,1628,1576,1470,1292,1191,987,761,693
(5)质子核磁共振光谱(400MHz,CDCl3):
δ=0.90(3H,t,J=6.6),1.23-1.71(10H,m),2.19(3H,s),2.84(2H,m),5.71(2H,s),7.38(1H,ddd,J=8.0,6.8,0.9),7.46(1H,d,J=8.7),7.68(1H,ddd,J=8.7,6.8,1.6),8.45(1H,dd,J=8.0,1.6)
(6)碳13核磁共振光谱(100MHz,CDCl3):
δ=11.60,14.05,22.56,28.59,28.88,29.76,30.55,31.66,56.26,114.42,118.19,123.83,124.60,127.31,132.41,139.86,141.68,149.60,177.64
<结构式(12)表示的化合物的理化性质>
作为所述结构式(12)表示的化合物的理化性质,如下。
(1)外观:黄色粉状
(2)熔点:167℃~170℃
(3)分子式:C20H28ON2S2
(4)高分辨质谱分析(HRESI-MS)(m/z):
实验值399.1534(M+Na)+
计算值399.1535(以C20H28ON2NaS2计)
(5)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:3119,2958,2918,2850,1619,1598,1538,1282,1199,1105,938,764,688
(6)质子核磁共振光谱(400MHz,CDCl3):
δ=0.86(3H,t,J=6.6),1.21-1.50(13H,m),2.22-2.58(2H,br),2.76(3H,s),5.68-6.41(2H,br),7.22(1H,t,J=7.8),7.44(1H,d,J=8.7),7.59(1H,ddd,J=8.7,7.8,1.1),8.15(1H,d,J=7.8),10.09(1H,br)
(7)碳13核磁共振光谱(100MHz,CDCl3):
δ=10.74,14.02,18.01,22.53,28.29,28.77,29.64,30.85,31.59,58.21,115.93,116.84,123.54,123.91,126.35,132.65,139.39,152.23,177.13,199.84
<结构式(13)表示的化合物的理化性质>
作为所述结构式(13)表示的化合物的理化性质,如下。
(1)外观:白色粉状
(2)熔点:92℃~94℃
(3)分子式:C21H30ON2S2
(4)高分辨质谱分析(HRESI-MS)(m/z):
实验值413.1689(M+Na)+
计算值413.1692(以C21H30ON2NaS2计)
(5)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:2958,2922,2852,1618,1595,1566,1492,1370,1277,1192,1004,769,700
(6)质子核磁共振光谱(400MHz,CDCl3):
δ=0.89(3H,t,J=6.8),1.24-1.50(8H,m),1.64(2H,m),2.22(3H,s),2.28(3H,s),2.71(3H,s),2.77(2H,m),5.60(2H,s),7.31(2H,m),7.55(1H,ddd,J=8.4,7.1,1.6),8.46(1H,dd,J=8.0,1.6)
(7)碳13核磁共振光谱(100MHz,CDCl3):
δ=11.50,14.06,14.75,15.03,22.61,28.28,28.92,29.83,30.66,31.76,63.75,115.84,116.97,122.77,124.81,126.75,131.27,141.06,151.35,161.39,177.54
所述化合物是否具有以所述结构式(1)~(13)中的任一个表示的结构能够通过适当选择的各种分析方法进行确认,例如可以举出所述质谱分析法、所述红外分光法、所述质子核磁共振分光法、所述碳13核磁共振分光法、紫外分光法等分析方法等。应予说明,由所述各分析方法而得的测定值有时会产生一些误差,但只要是本领域技术人员,就可以容易地鉴定出所述化合物具有以所述结构式(1)~(13)中的任一个表示的结构。
所述化合物可以是以所述结构式(1)~(13)中的任一个表示的化合物的盐。
作为所述盐,只要是可以在药理学上容许的盐,就没有特别限制,能够根据目的适当选择,例如可以举出乙酸盐、柠檬酸盐等有机盐、盐酸盐、碳酸盐等。
所述结构式(1)~(13)中的任一个表示的化合物可以为其互变异构体。
作为所述结构式(1)~(13)中的任一个表示的化合物的制造方法,没有特别限制,能够根据目的适当选择,但优选通过后述的本发明的制造方法制得。
<用途>
所述结构式(1)~(13)中的任一个表示的化合物具有优异的抗癌作用、或优异的抗幽门螺杆菌活性,是安全性高的化合物。因此,所述结构式(1)~(13)中的任一个表示的化合物例如可以优选用作后述的含本发明的化合物的组合物、本发明的抗癌剂、本发明的抗幽门螺杆菌剂等的有效成分。
(化合物的制造方法)
<所述结构式(3)、(4)、和(8)中的任一个表示的化合物的制造方法>
作为所述结构式(3)、(4)、和(8)中的任一个表示的化合物的制造方法,只要是使下述结构式(17)、(18)、和(19)中的任一个表示的化合物、与碱金属的氢氧化物和碱土类金属的氢氧化物中的至少一个进行反应的方法,就没有特别限制,能够根据目的适当选择。
所谓所述碱金属是指锂、钠、钾、铷、铯、钫。所谓所述碱土类金属,是指钙、锶、钡、镭。
作为所述碱金属的氢氧化物,没有特别限制,能够根据目的适当选择,例如可以举出氢氧化锂、氢氧化钠、氢氧化钾等。
作为所述碱土类金属的氢氧化物,没有特别限制,能够根据目的适当选择,例如可以举出氢氧化钡等。
所述碱金属的氢氧化物和所述碱土类的氢氧化物可以使用任一个,也可以将两者并用。
所述碱金属的氢氧化物可以单独使用一种,也可以将两种以上并用。另外,所述碱土类金属的氢氧化物可以单独使用一种,也可以将两种以上并用。
所述碱金属的氢氧化物和所述碱土类金属的氢氧化物之中,优选氢氧化钠。
下面,作为所述结构式(3)、(4)、和(8)中的任一个表示的化合物的制造方法的优选方式,将以氨基苄腈为起始物质的方式说明如下。
--结构式(14)表示的化合物的制造--
所述结构式(14)表示的化合物例如能够按以下方式制造。
氩气氛下使氨基苄腈溶解在无水四氢呋喃(以下有时称为“THF”)中,冰浴下滴加溴化乙基镁。在室温下使其搅拌12小时后,在冰浴下滴加10%盐酸水溶液。滴加完成后,在冰浴下加入氢氧化钠,将pH调到7。分离有机层,用二乙基醚萃取水层。合并有机层进行芒硝干燥,蒸馏除去溶剂。能够采用硅胶色层分离法(己烷:乙酸乙酯=6:1)纯化得到的残渣而获得结构式(14)表示的化合物。
--结构式(17)表示的化合物的制造--
所述结构式(17)表示的化合物例如能够按以下方式制造。
氩气氛下使所述结构式(14)表示的化合物溶解在二氯甲烷中,加入三乙基胺。进一步在冰浴下滴加作为酰氯的反-8-甲基-6-壬酰氯,在室温下搅拌。用0.1N盐酸停止反应,用二氯甲烷萃取,接着用饱和碳酸氢钠水溶液、食盐水洗涤。将合并的有机层进行芒硝干燥后,蒸馏除去溶剂。能够采用硅胶色层分离法(己烷:乙酸乙酯)纯化得到的残渣而获得结构式(17)表示的化合物。
-结构式(3)表示的化合物的制造-
以所述结构式(3)表示的化合物例如能够按以下方式制造。
在所述结构式(17)表示的化合物的二噁烷0.1M溶液中加入氢氧化钠,在110℃下搅拌1小时~2小时。将反应溶液恢复到室温,加入水,进一步加入1N盐酸直至pH变成7。进一步加入己烷施加超声波,则固体析出。抽滤固体,用水、己烷或己烷/乙酸乙酯=1:1的混合溶剂洗涤。能够洗涤后使其干燥,从而获得结构式(3)表示的化合物。
--结构式(18)表示的化合物的制造--
所述结构式(18)表示的化合物例如能够按以下方式制造。
氩气氛下使所述结构式(14)表示的化合物溶解在二氯甲烷中,加入三乙基胺。进一步在冰浴下滴加作为酰氯的异戊酰氯,在室温下搅拌。用0.1N盐酸停止反应,用二氯甲烷萃取,接着用饱和碳酸氢钠水溶液、食盐水洗涤。将合并的有机层进行芒硝干燥后,蒸馏除去溶剂。能够采用硅胶色层分离法(己烷:乙酸乙酯)纯化得到的残渣而获得结构式(18)表示的化合物。
-结构式(4)表示的化合物的制造-
所述结构式(4)表示的化合物例如能够按以下方式制造。
在所述结构式(18)表示的化合物的1,4-二噁烷0.1M溶液中加入氢氧化钠,在110℃下搅拌1小时~2小时。将反应溶液恢复到室温,加入水,进一步加入1N盐酸直至pH变成7。进一步加入己烷施加超声波,则固体析出。抽滤固体,用水、己烷或己烷/乙酸乙酯=1:1的混合溶剂洗涤。能够洗涤后使其干燥,从而获得结构式(4)表示的化合物。
--结构式(19)表示的化合物的制造--
所述结构式(19)表示的化合物例如能够按以下方式制造。
氩气氛下使所述结构式(14)表示的化合物溶解在二氯甲烷中,加入三乙基胺。进一步在冰浴下滴加作为酰氯的肉桂酰氯,在室温下搅拌。用0.1N盐酸停止反应,用二氯甲烷萃取,接着用饱和碳酸氢钠水溶液、食盐水洗涤。将合并的有机层进行芒硝干燥后,蒸馏除去溶剂。能够采用硅胶色层分离法(己烷:乙酸乙酯)纯化得到的残渣而获得结构式(19)表示的化合物。
-结构式(8)表示的化合物的制造-
所述结构式(8)表示的化合物例如能够按以下方式制造。
在所述结构式(19)表示的化合物的1,4-二噁烷0.1M溶液中加入氢氧化钠,在110℃下搅拌1小时~2小时。将反应溶液恢复到室温,加入水,进一步加入1N盐酸直至pH变成7。进一步加入己烷施加超声波,则固体析出。抽滤固体,用水、己烷或己烷/乙酸乙酯=1:1的混合溶剂洗涤。能够洗涤后使其干燥,从而获得结构式(8)表示的化合物。
<所述结构式(5)表示的化合物的制造方法>
作为所述结构式(5)表示的化合物的制造方法,只要是使下述结构式(21)表示的化合物、与碱金属的醇盐、碱金属的碳酸盐、和碱金属的氢化物中的至少一个反应后、使所述反应物与溴乙酸甲酯进行反应的方法,就没有特别限制,能够根据目的适当选择。
其中,所述结构式(5)中,Me表示甲基。
所谓所述碱金属,是指锂、钠、钾、铷、铯、钫。
作为所述碱金属的醇盐,没有特别限制,能够根据目的适当选择,例如可以举出叔丁醇锂等。
作为所述碱金属的碳酸盐,没有特别限制,能够根据目的适当选择,例如可以举出碳酸钠、碳酸钾、碳酸氢钠、碳酸氢钾等。
作为所述碱金属的氢化物,没有特别限制,能够根据目的适当选择,例如可以举出氢化钠。
所述碱金属的醇盐、所述碱金属的碳酸盐、和所述碱金属的氢化物可以单独使用任一种,也可以将两种以上并用。
所述碱金属的醇盐可以单独使用一种,也可以将两种以上并用。所述碱金属的碳酸盐可以单独使用一种,也可以将两种以上并用。另外,所述碱金属的氢化物可以单独使用一种,也可以将两种以上并用。
所述碱金属的醇盐、所述碱金属的碳酸盐、和所述碱金属的氢化物之中,优选叔丁醇锂、碳酸钾、氢化钠,更优选叔丁醇锂。
下面,作为以所述结构式(5)表示的化合物的制造方法的优选方式,将所述结构式(14)表示的化合物为起始物质的方式说明如下。
应予说明,所述结构式(14)表示的化合物能够优选根据上述方法制造。
--结构式(16)表示的化合物的制造--
所述结构式(16)表示的化合物例如能够按以下方式制造。
氩气氛下使所述结构式(14)表示的化合物溶解在二氯甲烷中,加入三乙基胺。进一步在冰浴下滴加作为酰氯的壬酰氯,在室温下搅拌。用0.1N盐酸停止反应,用二氯甲烷萃取,接着用饱和碳酸氢钠水溶液、食盐水洗涤。将合并的有机层进行芒硝干燥后,蒸馏除去溶剂。能够采用硅胶色层分离法(己烷:乙酸乙酯)纯化得到的残渣而获得结构式(16)表示的化合物。
--结构式(21)表示的化合物的制造--
所述结构式(21)表示的化合物例如能够按以下方式制造。
在所述结构式(16)表示的化合物的1,4-二噁烷0.1M溶液中加入氢氧化钠,在110℃下搅拌1小时~2小时。将反应溶液恢复到室温,加入水,进一步加入1N盐酸直至pH变成7。进一步加入己烷施加超声波,则固体析出。抽滤固体,用水、己烷或己烷/乙酸乙酯=1:1的混合溶剂洗涤。能够洗涤后使其干燥,从而获得结构式(21)表示的化合物。
-结构式(5)表示的化合物的制造-
所述结构式(5)表示的化合物例如能够按以下方式制造。
氩气氛下使所述结构式(21)表示的化合物溶解在THF中,加入叔丁醇锂的THF溶液在室温下搅拌20分钟。接着,加入溴乙酸甲酯,进一步回流下搅拌12小时。加入水,使反应停止,用乙酸乙酯萃取。对有机层进行芒硝干燥,蒸馏除去溶剂。能够将得到的残渣采用硅胶色层分离法(己烷:乙酸乙酯=2:1)纯化而获得结构式(5)表示的化合物。
<所述结构式(6)表示的化合物的制造方法>
作为所述结构式(6)表示的化合物的制造方法,只要是使下述结构式(5)表示的化合物、与碱金属的氢氧化物和碱土类金属的氢氧化物中的至少一个反应后、使所述反应物的pH成酸性的方法,就没有特别限制,能够根据目的适当选择。
其中,所述结构式(5)中,Me表示甲基。
所谓所述碱金属,是指锂、钠、钾、铷、铯、钫。所谓所述碱土类金属,是指钙、锶、钡、镭。
作为所述碱金属的氢氧化物,没有特别限制,能够根据目的适当选择,例如可以举出氢氧化锂、氢氧化钠、氢氧化钾等。
作为所述碱土类金属的氢氧化物,没有特别限制,能够根据目的适当选择,例如可以举出氢氧化钡等。
所述碱金属的氢氧化物和所述碱土类的氢氧化物可以使用任一个,也可以将两者并用。
所述碱金属的氢氧化物可以单独使用一种,也可以将两种以上并用。另外,所述碱土类金属的氢氧化物可以单独使用一种,也可以将两种以上并用。
所述碱金属的氢氧化物和所述碱土类金属的氢氧化物之中,优选氢氧化钠。
下面,作为所述结构式(6)表示的化合物的制造方法的优选方式,将以所述结构式(5)表示的化合物为起始物质的方式说明如下。
应予说明,所述结构式(5)表示的化合物能够优选根据上述方法制造。
-结构式(6)表示的化合物的制造-
所述结构式(6)表示的化合物例如能够按以下方式制造。
使所述结构式(5)表示的化合物溶解在乙醇(以下有时称为“EtOH”)与THF的混合溶剂中,加入2M氢氧化钠水溶液,在室温下搅拌2小时。冰浴下加入1N盐酸将pH调到4,用乙酸乙酯萃取。能够对有机层进行芒硝干燥,蒸馏除去溶剂,从而获得结构式(6)表示的化合物。
<所述结构式(1)表示的化合物的制造方法>
作为所述结构式(1)表示的化合物的制造方法,只要是使下述结构式(21)表示的化合物、与碱金属的碳酸盐和碱金属的氢化物中的至少一个、与溴乙酸甲酯进行反应的方法,就没有特别限制,能够根据目的适当选择。
其中,所述结构式(1)中,Me表示甲基。
所谓所述碱金属,是指锂、钠、钾、铷、铯、钫。
作为所述碱金属的碳酸盐,没有特别限制,能够根据目的适当选择,例如可以举出碳酸钠、碳酸钾、碳酸氢钠、碳酸氢钾等。
作为所述碱金属的氢化物,没有特别限制,能够根据目的适当选择,例如可以举出氢化钠。
所述碱金属的碳酸盐和所述碱金属的氢化物可以使用任一个,也可以将两者并用。
所述碱金属的碳酸盐可以单独使用一种,也可以将两种以上并用。另外,所述碱金属的氢化物可以单独使用一种,也可以将两种以上并用。
所述碱金属的碳酸盐和所述碱金属的氢化物之中,优选碳酸钾、氢化钠,更优选碳酸钾。
下面,作为所述结构式(1)表示的化合物的制造方法的优选方式,将以所述结构式(21)表示的化合物为起始物质的方式说明如下。
应予说明,所述结构式(21)表示的化合物能够优选根据上述方法制造。
-结构式(1)表示的化合物的制造-
所述结构式(1)表示的化合物例如能够按以下方式制造。
使所述结构式(21)溶解在N,N-二甲基甲酰胺(以下,有时称为“DMF”)中,加入碳酸钾、溴乙酸甲酯,在80℃下搅拌12小时。加入水,使反应停止,用乙酸乙酯萃取。对有机层进行芒硝干燥,蒸馏除去溶剂。能够将得到的残渣采用硅胶色层分离法(己烷:乙酸乙酯=2:1)纯化而获得结构式(1)表示的化合物。
<所述结构式(2)表示的化合物的制造方法>
作为所述结构式(2)表示的化合物的制造方法,只要是使下述结构式(1)表示的化合物、与碱金属的氢氧化物和碱土类金属的氢氧化物中的至少一个反应后、使所述反应物的pH成酸性的方法,就没有特别限制,能够根据目的适当选择。
其中,所述结构式(1)中,Me表示甲基。
所谓所述碱金属,是指锂、钠、钾、铷、铯、钫。所谓所述碱土类金属,是指钙、锶、钡、镭。
作为所述碱金属的氢氧化物,没有特别限制,能够根据目的适当选择,例如可以举出氢氧化锂、氢氧化钠、氢氧化钾等。
作为所述碱土类金属的氢氧化物,没有特别限制,能够根据目的适当选择,例如可以举出氢氧化钡等。
所述碱金属的氢氧化物和所述碱土类的氢氧化物可以使用任一个,也可以将两者并用。
所述碱金属的氢氧化物可以单独使用一种,也可以将两种以上并用。另外,所述碱土类金属的氢氧化物可以单独使用一种,也可以将两种以上并用。
所述碱金属的氢氧化物和所述碱土类金属的氢氧化物之中,优选氢氧化钠。
作为使所述反应物的pH成酸性的方法,没有特别限制,能够根据目的适当选择,例如可以举出1N盐酸等。
下面,作为所述结构式(2)表示的化合物的制造方法的优选方式,将以所述结构式(1)表示的化合物为起始物质的方式说明如下。
应予说明,所述结构式(1)表示的化合物能够优选根据上述方法制造。
-结构式(2)表示的化合物的制造-
所述结构式(2)表示的化合物例如能够按以下方式制造。
使所述结构式(1)表示的化合物溶解在EtOH与THF的混合溶剂中,加入2M氢氧化钠水溶液,在室温下搅拌2小时。冰浴下加入1N盐酸,将pH调到4,用乙酸乙酯萃取。能够对有机层进行芒硝干燥,蒸馏除去溶剂,从而获得结构式(2)表示的化合物。
<所述结构式(7)表示的化合物的制造方法>
作为所述结构式(7)表示的化合物的制造方法,只要是使下述结构式(21)表示的化合物、与碱金属的醇盐和碱金属的氢化物中的至少一个反应后、使所述反应物与溴化氰进行反应的方法,就没有特别限制,能够根据目的适当选择。
所谓所述碱金属,是指锂、钠、钾、铷、铯、钫。
作为所述碱金属的醇盐,没有特别限制,能够根据目的适当选择,例如可以举出叔丁醇锂等。
作为所述碱金属的氢化物,没有特别限制,能够根据目的适当选择,例如可以举出氢化钠。
所述碱金属的醇盐和所述碱金属的氢化物可以使用任一个,也可以将两者并用。
所述碱金属的醇盐可以单独使用一种,也可以将两种以上并用。另外,所述碱金属的氢化物可以单独使用一种,也可以将两种以上并用。
所述碱金属的醇盐和所述碱金属的氢化物之中,优选叔丁醇锂、氢化钠,更优选叔丁醇锂。
下面,作为所述结构式(7)表示的化合物的制造方法的优选方式,将所述结构式(21)表示的化合物为起始物质的方式说明如下。
应予说明,所述结构式(21)表示的化合物能够优选根据上述方法制造。
-结构式(7)表示的化合物的制造-
所述结构式(7)表示的化合物例如能够按以下方式制造。
使所述结构式(21)表示的化合物溶解在THF中,加入叔丁醇锂,在室温下搅拌20分钟。接着,加入溴化氰,在室温下搅拌2小时。加入水,使反应停止,用乙酸乙酯萃取。对有机层进行芒硝干燥,蒸馏除去溶剂。能够将得到的残渣采用硅胶色层分离法(己烷:乙酸乙酯=2:1)纯化,从而获得结构式(7)表示的化合物(34.0mg,62%)。
<所述结构式(9)表示的化合物的制造方法>
作为所述结构式(9)表示的化合物的制造方法,只要是使下述结构式(6)表示的化合物、与叔胺和吡啶类中的至少一个、与叠氮磷酸二苯酯、与甲硫醇钠进行反应的方法,就没有特别限制,能够根据目的适当选择。
其中,所述结构式(9)中、Me表示甲基。
作为所述叔胺,没有特别限制,能够根据目的适当选择,例如可以举出三乙基胺、N,N-二异丙基乙胺等。
作为所述吡啶类,没有特别限制,能够根据目的适当选择,例如可以举出吡啶、二甲基氨基吡啶等。
所述叔胺和所述吡啶类可以使用任一个,也可以将两者并用。
所述叔胺可以单独使用一种,也可以将两种以上并用。另外,所述吡啶类可以单独使用一种,也可以将两种以上并用。
所述叔胺和所述吡啶类之中,优选三乙基胺。
下面,作为所述结构式(9)表示的化合物的制造方法的优选方式,将以所述结构式(6)表示的化合物为起始物质的方式说明如下。
应予说明,所述结构式(6)表示的化合物能够优选根据上述方法制造。
-结构式(9)表示的化合物的制造-
所述结构式(9)表示的化合物例如能够按以下方式制造。
使所述结构式(6)表示的化合物悬浮于THF中,加入三乙基胺、叠氮磷酸二苯酯(以下有时称为“DPPA”)、甲硫醇钠,回流下搅拌2小时。加入氯化铵水溶液,用乙酸乙酯萃取。对有机层进行芒硝干燥,蒸馏除去溶剂。能够将得到的残渣采用硅胶色层分离法(己烷:乙酸乙酯=3:1)纯化而获得结构式(9)表示的化合物。
<所述结构式(10)表示的化合物的制造方法>
作为所述结构式(10)表示的化合物的制造方法,只要是使下述结构式(6)表示的化合物、与叔胺和吡啶类中的至少一个、与叠氮磷酸二苯酯反应后、使所述反应物与甲硫醇钠进行反应的方法,就没有特别限制,能够根据目的适当选择。
其中,所述结构式(10)中,Me表示甲基。
作为所述叔胺,没有特别限制,能够根据目的适当选择,例如可以举出三乙基胺、N,N-二异丙基乙胺等。
作为所述吡啶类,没有特别限制,能够根据目的适当选择,例如可以举出吡啶、二甲基氨基吡啶等。
所述叔胺和所述吡啶类可以使用任一个,也可以将两者并用。
所述叔胺可以单独使用一种,也可以将两种以上并用。另外,所述吡啶类可以单独使用一种,也可以将两种以上并用。
所述叔胺和所述吡啶类之中,优选三乙基胺。
下面,作为所述结构式(10)表示的化合物的制造方法的优选方式,将以所述结构式(6)表示的化合物为起始物质的方式说明如下。
应予说明,所述结构式(6)表示的化合物能够优选根据上述方法制造。
-结构式(10)表示的化合物的制造-
所述结构式(10)表示的化合物例如能够按以下方式制造。
使所述结构式(6)表示的化合物悬浮在THF中,在冰冷却下加入三乙基胺、DPPA,进一步回流下搅拌1小时。在其中加入甲硫醇钠,进一步搅拌1小时。加入氯化铵水溶液,用乙酸乙酯萃取。对有机层进行芒硝干燥,蒸馏除去溶剂。能够将得到的残渣采用硅胶色层分离法(己烷:乙酸乙酯=5:1)纯化而获得结构式(10)表示的化合物。
<所述结构式(11)表示的化合物的制造方法>
作为所述结构式(11)表示的化合物的制造方法,只要是使下述结构式(20)表示的化合物、与碱金属的醇盐和碱金属的氢化物中的至少一个反应后、使所述反应物与硫氰酸氯甲酯进行反应的方法,就没有特别限制,能够根据目的适当选择。
所谓所述碱金属,是指锂、钠、钾、铷、铯、钫。
作为所述碱金属的醇盐,没有特别限制,能够根据目的适当选择,例如可以举出叔丁醇锂等。
作为所述碱金属的氢化物,没有特别限制,能够根据目的适当选择,例如可以举出氢化钠。
所述碱金属的醇盐和所述碱金属的氢化物可以使用任一个,也可以将两者并用。
所述碱金属的醇盐可以单独使用一种,也可以将两种以上并用。另外,所述碱金属的氢化物可以单独使用一种,也可以将两种以上并用。
所述碱金属的醇盐和所述碱金属的氢化物之中,优选叔丁醇锂、氢化钠,更优选叔丁醇锂。
下面,作为所述结构式(11)表示的化合物的制造方法的优选方式,将以所述结构式(14)表示的化合物为起始物质的方式说明如下。
应予说明,所述结构式(14)表示的化合物能够优选根据上述方法制造。
--结构式(15)表示的化合物的制造--
所述结构式(15)表示的化合物例如能够按以下方式制造。
氩气氛下使所述结构式(14)表示的化合物溶解在二氯甲烷中,加入三乙基胺。进一步在冰浴下滴加作为酰氯的辛酰氯,在室温下搅拌。用0.1N盐酸停止反应,用二氯甲烷萃取,接着用饱和碳酸氢钠水溶液、食盐水洗涤。将合并的有机层进行芒硝干燥后,蒸馏除去溶剂。能够采用硅胶色层分离法(己烷:乙酸乙酯)纯化得到的残渣而获得结构式(15)表示的化合物。
--结构式(20)表示的化合物的制造--
所述结构式(20)表示的化合物例如能够按以下方式制造。
在所述结构式(15)表示的化合物的二噁烷0.1M溶液中加入氢氧化钠,在110℃下搅拌1小时~2小时。将反应溶液恢复到室温,加入水,进一步加入1N盐酸直至pH变成7。进一步加入己烷施加超声波,则固体析出。抽滤固体,用水、己烷或己烷/乙酸乙酯=1:1的混合溶剂洗涤。能够洗涤后使其干燥,从而获得结构式(20)表示的化合物。
-结构式(11)表示的化合物的制造-
所述结构式(11)表示的化合物例如能够按以下方式制造。
氩气氛下使所述结构式(20)表示的化合物溶解在THF中,加入叔丁醇锂的THF溶液,在室温下搅拌20分钟。冰冷却下在其中滴加硫氰酸氯甲酯,进一步在室温下搅拌2小时。加入食盐水使反应停止,用乙酸乙酯萃取。能够对有机层进行芒硝干燥后,采用硅胶色层分离法(己烷:乙酸乙酯=2:1)纯化得到的残渣,从而获得结构式(11)表示的化合物。
<所述结构式(12)表示的化合物的制造方法>
作为所述结构式(12)表示的化合物的制造方法,只要是在乙腈的存在下使下述结构式(11)表示的化合物与甲硫醇钠进行反应的方法,就没有特别限制,能够根据目的适当选择。
其中,所述结构式(12)中,Me表示甲基。
下面,作为所述结构式(12)表示的化合物的制造方法的优选方式,将以所述结构式(11)表示的化合物为起始物质的方式说明如下。
应予说明,所述结构式(11)表示的化合物能够优选根据上述方法制造。
-结构式(12)表示的化合物的制造-
所述结构式(12)表示的化合物例如能够按以下方式制造。
在所述结构式(11)表示的化合物与甲硫醇钠的混合物中加入乙腈,在室温下搅拌20分钟。加入饱和碳酸氢钠水溶液,用乙酸乙酯萃取。能够对有机层进行芒硝干燥后,采用硅胶色层分离法(己烷:乙酸乙酯=2:1)纯化得到的残渣,从而获得结构式(12)表示的化合物。
<所述结构式(13)表示的化合物的制造方法>
作为所述结构式(13)表示的化合物的制造方法,只要是在乙腈的存在下使下述结构式(11)表示的化合物与甲硫醇钠反应后、使所述反应物与甲基化试剂进行反应的方法,就没有特别限制,能够根据目的适当选择。
其中,所述结构式(13)中,Me表示甲基。
作为所述甲基化试剂,没有特别限制,能够根据目的适当选择,例如可以举出碘代甲烷、三氟甲磺酸甲酯、硫酸二甲酯、Meerwein试剂等。所述甲基化试剂可以单独使用一种,也可以将两种以上并用。这些之中,优选碘代甲烷。
下面,作为所述结构式(13)表示的化合物的制造方法的优选方式,将以所述结构式(11)表示的化合物为起始物质的方式说明如下。
应予说明,所述结构式(11)表示的化合物能够优选根据上述方法制造。
-结构式(13)表示的化合物的制造-
所述结构式(13)表示的化合物例如能够按以下方式制造。
在所述结构式(11)表示的化合物与甲硫醇钠的混合物中加入乙腈,在室温下搅拌20分钟。接着,在室温下加入碘甲烷,进一步搅拌30分钟。加入饱和碳酸氢钠水溶液,用乙酸乙酯萃取。能够对有机层进行芒硝干燥后,采用硅胶色层分离法(己烷:乙酸乙酯=2:1)纯化得到的残渣,从而获得结构式(13)表示的化合物。
对于所述各结构式表示的化合物的制造方法中的反应条件、使用的化合物及其用量、溶剂等,只要不损害本发明的效果,就没有特别限制,能够根据目的适当选择。
所述各化合物是否具有所述各结构式表示的结构能够通过适当选择的各种分析方法来确认,例如可以举出所述质谱分析法、所述紫外分光法、所述红外分光法、所述质子核磁共振分光法、所述碳13核磁共振分光法等分析方法等。
(含化合物的组合物、抗癌剂、抗幽门螺杆菌剂)
<含化合物的组合物>
本发明的含化合物的组合物至少含有所述结构式(1)~(13)中的任一个表示的化合物,根据需要地进一步含有其它成分。
所述结构式(1)~(13)中的任一个表示的化合物可以单独使用一种,也可以将两种以上并用。
作为所述含化合物的组合物中的以所述结构式(1)~(13)中的任一个表示的化合物的含量,没有特别限制,能够根据目的适当选择。所述含化合物的组合物可以是以所述结构式(1)~(13)中的任一个表示的化合物其自身。
-其它成分-
作为所述其它成分,没有特别限制,能够从可在药理学上容许的载体之中根据目的适当选择,例如可以举出添加剂、辅剂、水等。这些可以单独使用一种,也可以将两种以上并用。
作为所述添加剂或所述辅剂,没有特别限制,能够根据目的适当选择,例如可以举出杀菌剂、保鲜剂、粘结剂、增稠剂、固定剂、粘合剂、着色剂、稳定化剂、pH调节剂、缓冲剂、等渗化剂、溶剂、抗氧化剂、防紫外线剂、防晶析剂、消泡剂、物性提高剂、防腐剂等。
作为所述杀菌剂,没有特别限制,能够根据目的适当选择,例如可以举出氯化苄烷铵、苯索氯铵、十六烷基氯化吡啶鎓等阳离子性表面活性剂等。
作为所述保鲜剂,没有特别限制,能够根据目的适当选择、例如可以举出对羟基苯甲酸酯类、氯代丁醇、甲酚等。
作为所述粘结剂、增稠剂、固定剂,没有特别限制,能够根据目的适当选择,例如可以举出、淀粉、糊精、纤维素、甲基纤维素、乙基纤维素、羧甲基纤维素、羟基乙基纤维素、羟基丙基纤维素、羟基丙基甲基纤维素、羧甲基淀粉、支链淀粉、海藻酸钠、海藻酸铵、海藻酸丙二醇酯、瓜耳胶、刺槐豆胶、阿拉伯树胶、黄原酸胶、凝胶、酪蛋白、聚乙烯醇、聚环氧乙烷、聚乙二醇、乙烯·丙烯嵌段聚合物、聚丙烯酸钠、聚乙烯吡咯烷酮等。
作为所述粘合剂,没有特别限制,能够根据目的适当选择,例如可以举出水、乙醇、丙醇、单糖浆、葡萄糖液、淀粉液、凝胶液、羧甲基纤维素、羟基丙基纤维素、羟基丙基淀粉、甲基纤维素、乙基纤维素、紫胶、磷酸钙、聚乙烯吡咯烷酮等。
作为所述着色剂,没有特别限制,能够根据目的适当选择,例如可以举出氧化钛、氧化铁等。
作为所述稳定化剂,没有特别限制,能够根据目的适当选择,例如可以举出黄蓍、阿拉伯树胶、凝胶、焦亚硫酸钠、乙二胺四乙酸(EDTA)、巯基乙酸、硫羟乳酸等。
作为所述pH调节剂或所述缓冲剂,没有特别限制,能够根据目的适当选择,例如可以举出柠檬酸钠、乙酸钠、磷酸钠等。
作为所述等渗化剂,没有特别限制,能够根据目的适当选择,例如可以举出氯化钠、葡萄糖等。
作为所述含化合物的组合物中的所述其它成分的含量,没有特别限制,能够在不损害所述结构式(1)~(13)中的任一个表示的化合物的效果的范围内,根据目的适当选择。
-用途-
所述含化合物的组合物含有所述结构式(1)~(13)中的任一个表示的化合物,因此,具有优异的抗癌作用、优异的抗幽门螺杆菌活性,安全性高,例如可优选用于药品组合物、抗癌剂、抗幽门螺杆菌剂等。
应予说明,所述含化合物的组合物可以单独使用,也可以与以其它成分为有效成分的药品合并使用。另外,所述含化合物的组合物可以以配合在以其它成分为有效成分的药品中的状态使用。
<抗癌剂>
本发明的抗癌剂至少含有所述结构式(1)~(13)中的任一个表示的化合物,根据需要地进一步含有其它成分。
所述结构式(1)~(13)中的任一个表示的化合物可以单独使用一种,也可以将两种以上并用。
作为所述抗癌剂中的所述结构式(1)~(13)中的任一个表示的化合物的含量,没有特别限制,能够根据目的适当选择。所述抗癌剂可以为所述结构式(1)~(13)中的任一个表示的化合物其自身。
-其它成分-
作为所述其它成分,没有特别限制,能够根据目的适当选择,例如可以举出与在所述含化合物的组合物中记载的其它成分相同的成分。这些可以单独使用一种,也可以将两种以上并用。
作为所述抗癌剂中的所述其它成分的含量,没有特别限制,能够在不损害所述结构式(1)~(13)中的任一个表示的化合物的效果的范围内,根据目的适当选择。
-用途-
所述抗癌剂含有所述结构式(1)~(13)中的任一个表示的化合物,因此,具有优异的抗癌作用,安全性高,可优选用作胃癌、前列腺癌、肺癌、大肠癌、胰腺癌、乳腺癌等广泛的癌的预防剂或治疗剂。这些之中,可特别优选用于胃癌、大肠癌。
应予说明,所述抗癌剂可以单独使用,也可以与以其它成分为有效成分的药品合并使用。另外,所述抗癌剂可以以配合在以其它成分为有效成分的药品中的状态使用。
另外,如后述的试验例所示,本发明的所述结构式(1)~(13)表示的化合物能够在正常间质细胞的存在下更加抑制癌细胞的增殖。
所述抗癌剂含有所述结构式(1)~(13)中的任一个表示的化合物,因此,通过对个体给药,能够预防个体的癌生成或者治疗患癌个体。所以,本发明也涉及以对个体给予所述抗癌剂为特征的癌的预防或治疗方法。
<抗幽门螺杆菌剂>
本发明的抗幽门螺杆菌剂至少含有所述结构式(1)~(13)中的任一个表示的化合物,根据需要进一步含有其它成分。
所述结构式(1)~(13)中的任一个表示的化合物可以单独使用一种,也可以将两种以上并用。
作为所述抗幽门螺杆菌剂中的所述结构式(1)~(13)中的任一个表示的化合物的含量,没有特别限制,能够根据目的适当选择。所述抗幽门螺杆菌剂可以为所述结构式(1)~(13)中的任一个表示的化合物其自身。
-其它成分-
作为所述其它成分,没有特别限制,能够根据目的适当选择,例如可以举出与在所述含化合物的组合物中记载的其它成分相同的成分。这些可以单独使用一种,也可以将两种以上并用。
作为所述抗幽门螺杆菌剂中的所述其它成分的含量,没有特别限制,能够在不损害所述结构式(1)~(13)中的任一个表示的化合物的效果的范围内,根据目的适当选择。
-用途-
所述抗幽门螺杆菌剂含有所述结构式(1)~(13)中的任一个表示的化合物,因此,具有优异的抗幽门螺杆菌活性,安全性高,可以优选用作胃溃疡、十二指肠溃疡等由幽门螺杆菌引起的胃和十二指肠损害的预防剂或治疗剂。
应予说明,所述抗幽门螺杆菌剂可以单独使用,也可以与以其它成分为有效成分的药品合并使用。另外,所述抗幽门螺杆菌剂可以以配合在以其它成分为有效成分的药品中的状态使用。
所述抗幽门螺杆菌剂含有以所述结构式(1)~(13)中的任一个表示的化合物,因此,通过对个体给药,能够预防个体感染幽门螺杆菌、或治疗感染幽门螺杆菌的个体。所以,本发明也涉及以对个体给予所述抗幽门螺杆菌剂为特征的由幽门螺杆菌带来的感染病的预防或治疗方法。
另外,通过将所述抗幽门螺杆菌剂对个体给药,能够预防由幽门螺杆菌引起的胃和十二指肠损害的发生、或治疗患有由幽门螺杆菌引起的胃和十二指肠损害的个体。所以,本发明也涉及以对个体给予所述抗幽门螺杆菌剂为特征的由幽门螺杆菌引起的胃和十二指肠损害的预防或治疗方法。
<剂型>
作为所述含化合物的组合物、抗癌剂、和抗幽门螺杆菌剂的剂型,没有特别限制,能够根据目的适当选择,例如可以举出固体剂、半固体剂、液体剂等。这些剂型的所述含化合物的组合物、抗癌剂、和抗幽门螺杆菌剂能够依据常规方法制造。
-固体剂-
作为所述固体剂,没有特别限制,能够根据目的适当选择,用作内用剂的情况下,例如可以举出片剂、咀嚼片、泡腾片、口腔崩解片、糖锭、滴剂、硬胶囊剂、软胶囊剂、颗粒剂、粉剂、丸剂、干糖浆剂、浸剂等。
所述固体剂用作外用剂的情况下,例如可以举出栓剂、泥敷剂、硬膏剂等。
-半固体剂-
作为所述半固体剂,没有特别限制,能够根据目的适当选择,用作内用剂的情况下,例如可以举出舐剂(煎膏剂)、咀嚼胶剂、蓬松胶剂(whipping agent)、胶冻剂等。
所述半固体剂用作外用剂的情况下,例如可以举出软膏剂、乳膏剂、摩丝剂、吸入剂、Nazar凝胶剂等。
-液体剂-
作为所述液体剂,没有特别限制,能够根据目的适当选择,用作内用剂的情况下,例如可以举出糖浆剂、饮剂、悬浮剂、醑剂等。
所述液体剂用作外用剂的情况下,例如可以举出液剂、滴眼剂、气雾剂、喷雾剂等。
<给药>
作为所述含化合物的组合物、抗癌剂、和抗幽门螺杆菌剂的给药方法、给药量、给药时间、和给药对象,没有特别限制,能够根据目的适当选择。
作为所述给药方法,例如可以举出局部给药法、肠道给药法、非口服给药法等。
作为所述给药量,没有特别限制,能够考虑给药对象个体的年龄、体重、体质、症状、是否给予以其它成分为有效成分的药品、药剂等各种各样的要因来适当选择。
作为成为所述给药对象的动物种类,没有特别限制,能够根据目的适当选择、例如可以举出人、猴、猪、牛、绵羊、山羊、狗、猫、小鼠、大鼠、鸟等,这些之中优选用于人。
实施例
下面,列举本发明的制造例、试验例对本发明具体地进行说明,但本发明不受这些制造例、试验例任何限定。应予说明,下述试验例的数据是获得了相同的结果、独立进行了两次或三次的实验的代表性数据。统计分析采用学生t检验(Student's t-test)。
(制造例1)
<结构式(3)表示的化合物的制造>
按以下方式,通过化学合成制造所述结构式(3)表示的化合物。
-结构式(14)表示的化合物的制造-
氩气氛下使氨基苄腈(15.0g,128mmol)溶解在无水四氢呋喃(以下有时称为“THF”)300mL中,冰浴下滴加溴化乙基镁(127mL,383mmol)。在室温下使其搅拌12小时后,在冰浴下滴加10%盐酸水溶液(100mL)。滴加完成后,在冰浴下加入氢氧化钠,将pH调到7。分离有机层,用二乙基醚萃取水层。合并有机层进行芒硝干燥,蒸馏除去溶剂。将得到的残渣采用硅胶色层分离法(己烷:乙酸乙酯=6:1)纯化,获得结构式(14)表示的化合物(9.7g,51%)。
--理化性质--
作为所述结构式(14)表示的化合物的理化性质,如下。
(1)外观:黄色粉末
(2)熔点:42℃~43℃
(3)分子式:C9H11ON
(4)高分辨质谱分析(HRESI-MS)(m/z):
实验值150.0911(M+H)+
计算值150.0913(以C9H12ON计)
(5)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:3434,3331,1644,1620
(6)质子核磁共振光谱(400MHz,CDCl3):
δ=1.21(3H,q,J=7.3),2.98(2H,q,J=7.3),6.27(2H,brs),6.62-6.67(2H,m),7.25(1H,ddd,J=7.3,5.0,1.4),7.76(1H,dd,J=8.7,1.4)
(7)碳13核磁共振光谱(100MHz,CDCl3):
δ=8.70,32.27,115.70,117.30,117.84,131.10,134.05,150.21,203.33
-结构式(17)表示的化合物的制造-
氩气氛下使所述结构式(14)表示的化合物(1当量)溶解在二氯甲烷中,加入三乙基胺(2当量)。进一步在冰浴下滴加作为酰氯的反-8-甲基-6-壬酰氯(1.1当量),在室温下使其搅拌。用0.1N盐酸停止反应,用二氯甲烷萃取,接着,用饱和碳酸氢钠水溶液、食盐水洗涤。合并有机层进行芒硝干燥,蒸馏除去溶剂。能够采用硅胶色层分离法(己烷:乙酸乙酯)纯化得到的残渣而获得结构式(17)表示的化合物。
--理化性质--
作为所述结构式(17)表示的化合物的理化性质,如下。
(1)外观:无色油状物
(2)分子式:C19H27O2N
(3)高分辨质谱分析(HRESI-MS)(m/z):
实验值324.1932(M+Na)+
计算值324.1934(以C19H27O2NNa计)
(4)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:3255,2956,2937,1655,969,754
(5)质子核磁共振光谱(400MHz,CDCl3):
δ=0.95(6H,d,J=6.8),1.22(3H,t,J=7.1),1.44(2H,m),1.75(2H,m),2.02(2H,m),2.22(1H,m),2.44(2H,t,J=7.3),3.07(2H,q,J=7.1),5.37(2H,m),7.09(1H,ddd,J=8.0,7.3,1.1),7.53(1H,ddd,J=8.5,7.3,1.4),7.92(1H,dd,J=8.0,1.4),8.77(1H,dd,J=8.5,1.1),11.77(1H,brs)
(6)碳13核磁共振光谱(100MHz,CDCl3):
δ=8.45,22.64,25.03,29.17,30.96,32.21,33.14,38.66,120.84,121.38,122.14,126.50,130.65,134.84,138.03,141.06,172.68,205.36
-结构式(3)表示的化合物的制造-
在所述结构式(17)表示的化合物(1当量)的1,4-二噁烷0.1M溶液中加入氢氧化钠(3.0当量),在110℃下使其搅拌1小时~2小时。将反应溶液恢复到室温,加入水,进一步加入1N盐酸直至pH变成7。进一步加入己烷施加超声波,则固体析出。抽滤固体,依次用水、己烷、接着用己烷/乙酸乙酯=1∶1的混合溶剂洗涤。能够洗涤后使其干燥,从而获得结构式(3)表示的化合物。
--理化性质--
作为所述结构式(3)表示的化合物的理化性质,如下。
(1)外观:白色粉状
(2)熔点:178℃~181℃
(3)分子式:C19H25ON
(4)高分辨质谱分析(HRESI-MS)(m/z):
实验值284.2011(M+H)+
计算值284.2009(以C19H26ON计)
(5)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:3064,2957,2933,1670,1638,1614,1555,1500,1371,1358,1152,1028,998,967,756,691
(6)质子核磁共振光谱(600MHz,CDCl3):
δ=0.95(6H,d,J=6.5),1.44(2H,m),1.69(2H,m),2.01(2H,q,J=6.8),2.15(3H,s),2.21(1H,m),2.70(2H,m),5.27-5.32(1H,m),5.36-5.39(1H,m),7.28(1H,ddd,J=8.2,5.8,1.0),7.32(1H,brd,J=8.2),7.52(1H,ddd,J=8.2,5.5,1.4),8.36(1H,dd,J=8.2,1.4),8.65(1H,br)
(7)碳13核磁共振光谱(150MHz,CDCl3):
δ=10.65,22.64,27.77,29.22,30.98,32.09,32.96,115.72,116.69,123.00,123.66,126.14,126.30,131.25,138.49,138.78,148.54,178.16
(制造例2)
<结构式(4)表示的化合物的制造>
按以下方式,通过化学合成制造所述结构式(4)表示的化合物。
-结构式(18)表示的化合物的制造-
氩气氛下使所述结构式(14)表示的化合物(1当量)溶解在二氯甲烷中,加入三乙基胺(2当量)。进一步在冰浴下滴加作为酰氯的异戊酰氯(1.1当量),在室温下使其搅拌。用0.1N盐酸停止反应,用二氯甲烷萃取,接着,用饱和碳酸氢钠水溶液、食盐水洗涤。合并有机层进行芒硝干燥,蒸馏除去溶剂。能够采用硅胶色层分离法(己烷∶乙酸乙酯)纯化得到的残渣而获得结构式(18)表示的化合物。
--理化性质--
作为所述结构式(18)表示的化合物的理化性质,如下。
(1)外观:无色油状物
(2)分子式:C14H19O2N
(3)高分辨质谱分析(HRESI-MS)(m/z):
实验值256.1306(M+Na)+
计算值256.1308(以C14H19O2NNa计)
(4)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:3254,2959,1697,1376,754
(5)质子核磁共振光谱(400MHz,CDCl3):
δ=1.01(6H,d,J=6.4),1.22(3H,t,J=7.1),2.24(1H,m),2.31(2H,d,J=6.4),3.07(2H,q,J=7.1),7.10(1H,ddd,J=8.4,6.8,1.1),7.53(1H,ddd,J=8.5,6.8,1.4),7.93(1H,dd,J=8.0,1.4),8.78(1H,d,J=8.5),11.75(1H,brs)
(6)碳13核磁共振光谱(100MHz,CDCl3):
δ=8.45,22.46,26.20,33.15,48.09,120.81,121.38,122.16,130.59,134.85,141.02,172.14,205.40
-结构式(4)表示的化合物的制造-
在所述结构式(18)表示的化合物(1当量)的1,4-二噁烷0.1M溶液中加入氢氧化钠(3.0当量),在110℃下使其搅拌1小时~2小时。将反应溶液恢复到室温,加入水,进一步加入1N盐酸直至pH变成7。进一步加入己烷施加超声波,则固体析出。抽滤固体,依次用水、己烷、接着用己烷/乙酸乙酯=1∶1的混合溶剂洗涤。能够洗涤后使其干燥,从而获得结构式(4)表示的化合物。
--理化性质--
作为所述结构式(4)表示的化合物的理化性质,如下。
(1)外观:白色粉状
(2)熔点:240℃~244℃;
(3)分子式:C14H17ON
(4)高分辨质谱分析(HRESI-MS)(m/z):
实验值216.1385(M+H)+
计算值216.1383(以C14H18ON计)
(5)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:3059,2956,1636,1609,1554,1505,1369,1359,1189,998,762,695
(6)质子核磁共振光谱(400MHz,DMSO-d6):
δ=0.89(6H,d,J=6.6),1.94(3H,s),1.99(1H,m),2.53(2H,d,J=7.5),7.18(1H,ddd,J=8.2,6.6,1.4),7.46(1H,d,J=8.2),7.52(1H,ddd,J=8.2,6.6,1.4),8.01(1H,dd,J=8.2,1.1),11.23(1H,br s)
(7)碳13核磁共振光谱(100MHz,DMSO-d6):
δ=11.03,22.28,28.30,114.68,117.74,122.39,123.00,125.18,131.08,139.33,148.76,176.43
(制造例3)
<结构式(8)表示的化合物的制造>
按以下方式,通过化学合成制造所述结构式(8)表示的化合物。
-结构式(19)表示的化合物的制造-
氩气氛下使所述结构式(14)表示的化合物(1当量)溶解在二氯甲烷中,加入三乙基胺(2当量)。进一步在冰浴下滴加作为酰氯的肉桂酰氯(1.1当量),在室温下使其搅拌。用0.1N盐酸停止反应,用二氯甲烷萃取,接着,用饱和碳酸氢钠水溶液、食盐水洗涤。合并有机层进行芒硝干燥,蒸馏除去溶剂。能够采用硅胶色层分离法(己烷∶乙酸乙酯)纯化得到的残渣而获得结构式(19)表示的化合物。
--理化性质--
作为所述结构式(19)表示的化合物的理化性质,如下。
(1)外观:白色粉状
(2)熔点:109℃~111℃
(3)分子式:C18H17O2N
(4)高分辨质谱分析(HRESI-MS)(m/z):
实验值302.1151(M+Na)+
计算值302.1152(以C18H17O2NNa计)
(5)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:3223,3023,1677,1653,751,727
(6)质子核磁共振光谱(400MHz,CDCl3):
δ=1.25(3H,t,J=7.1),3.11(2H,q,J=7.1),6.65(1H,d,J=15.5),7.13(1H,ddd,J=8.0,7.1,1.1),7.36-7.44(3H,m),7.56-7.62(3H,m),7.76(1H,d,J=15.5),7.96(1H,dd,J=8.0,1.4),8.91(1H,dd,J=8.5,1.4),12.10(1H,brs)
(7)碳13核磁共振光谱(100MHz,CDCl3):
δ=8.43,33.15,121.06,121.47,122.09,122.41,128.07,128.83,129.95,130.69,134.66,134.93,141.25,142.25,164.85,205.56
-结构式(8)表示的化合物的制造-
在所述结构式(19)表示的化合物(1当量)的1,4-二噁烷0.1M溶液中加入氢氧化钠(3.0当量),在110℃下使其搅拌1小时~2小时。将反应溶液恢复到室温,加入水,进一步加入1N盐酸直至pH变成7。进一步加入己烷施加超声波,则固体析出。抽滤固体,依次用水、己烷洗涤。能够洗涤后使其干燥,从而获得结构式(8)表示的化合物。
--理化性质--
作为所述结构式(8)表示的化合物的理化性质,如下。
(1)外观:黄色粉状
(2)熔点:>260℃
(3)分子式:C18H15ON
(4)高分辨质谱分析(HRESI-MS)(m/z):
实验值284.1046(M+Na)+
计算值284.1046(以C18H15ONNa计)
(5)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:3064,2938,1628,1570,1507,1387,1359,1187,965,755,690
(6)质子核磁共振光谱(400MHz,DMSO-d6):
δ=2.13(3H,s),7.21(1H,ddd,J=8.5,6.8,1.1),7.32-7.50(5H,m),7.55-7.59(1H,m),7.67-7.72(3H,m),8.03(1H,dd,J=8.2,1.4),11.20(1H,s)
(7)碳13核磁共振光谱(100MHz,DMSO-d6):
δ=10.66,115.68,118.18,121.14,122.55,123.13,125.14,127.57,129.16,129.30,131.61,135.11,135.94,139.75,143.14,176.76
(制造例4)
<结构式(5)表示的化合物的制造>
按以下方式,通过化学合成制造所述结构式(5)表示的化合物。
-结构式(16)表示的化合物的制造-
氩气氛下使所述结构式(14)表示的化合物(1当量)溶解在二氯甲烷中,加入三乙基胺(2当量)。进一步在冰浴下滴加作为酰氯的壬酰氯(1.1当量),在室温下使其搅拌。用0.1N盐酸停止反应、用二氯甲烷萃取,接着,用饱和碳酸氢钠水溶液、食盐水洗涤。合并有机层进行芒硝干燥,蒸馏除去溶剂。能够采用硅胶色层分离法(己烷∶乙酸乙酯)纯化得到的残渣而获得结构式(16)表示的化合物。
--理化性质--
作为所述结构式(16)表示的化合物的理化性质,如下。
(1)外观:无色油状物
(2)分子式:C18H27O2N
(3)高分辨质谱分析(HRESI-MS)(m/z):
实验值312.1933(M+Na)+
计算值312.1934(以C18H27O2NNa计)
(4)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:3255,2953,2928,1698,754
(5)质子核磁共振光谱(400MHz,CDCl3):
δ=0.87(3H,t,J=6.6),1.20-1.41(13H,m),1.74(2H,m),2.43(2H,t,J=7.6),3.08(2H,q,J=7.3),7.09(1H,ddd,J=8.0,7.3,1.1),7.53(1H,ddd,J=8.5,7.3,1.6),7.93(1H,dd,J=8.0,1.6),8.77(1H,dd,J=8.5,1.1),11.76(1H,br s)
(6)碳13核磁共振光谱(100MHz,CDCl3):
δ=8.54,14.07,22.63,25.55,29.14,29.22,29.28,31.87,33.20,38.80,120.85,121.96,122.12,130.57,134.84,141.08,172.80,205.37
-结构式(21)表示的化合物的制造-
在所述结构式(16)表示的化合物(1当量)的1,4-二噁烷0.1M溶液中加入氢氧化钠(3.0当量),在110℃下使其搅拌1小时~2小时。将反应溶液恢复到室温,加入水,进一步加入1N盐酸直至pH变成7。进一步加入己烷施加超声波,则固体析出。抽滤固体,用水、己烷洗涤。能够洗涤后使其干燥,从而获得结构式(21)表示的化合物。
--理化性质--
作为所述结构式(21)表示的化合物的理化性质,如下。
(1)外观:白色粉状
(2)熔点:228℃~231℃
(3)分子式:C18H25ON
(4)高分辨质谱分析(HRESI-MS)(m/z):
实验值272.2010(M+H)+
计算值272.2009(以C18H26ON计)
(5)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:3059,2952,2923,1638,1607,1555,1500,1190,998,754,693
(6)质子核磁共振光谱(400MHz,DMSO-d6):
δ=0.80(3H,t,J=6.6),1.19-1.35(10H,m),1.59(2H,m),1.94(3H,s),2.62(2H,t,J=7.5),7.18(1H,dd,J=8.2,6.6),7.45(1H,d,J=8.0),7.51(1H,ddd,J=8.2,6.7,1.4),8.00(1H,dd,J=8.0,1.4),11.31(1H,brs)
(7)碳13核磁共振光谱(100MHz,DMSO-d6):
δ=10.48,14.15,22.26,28.49,28.80,28.92,29.04,31.43,31.84,113.87,117.73,122.39,123.06,125.18,131.05,139.36,149.82,176.39
-结构式(5)表示的化合物的制造-
氩气氛下使所述结构式(21)表示的化合物(800mg,2.95mmol)溶解在THF(20mL)中,加入叔丁醇锂的THF溶液(4.4mL,4.42mmol),在室温下使其搅拌20分钟。接着,加入溴乙酸甲酯(1.4mL、14.7mmol),进一步使其回流下搅拌12小时。加入水,使反应停止,用乙酸乙酯萃取。对有机层进行芒硝干燥,蒸馏除去溶剂。将得到的残渣采用硅胶色层分离法(己烷∶乙酸乙酯=2∶1)纯化,从而获得结构式(5)表示的化合物(780mg,76%)。
--理化性质--
作为所述结构式(5)表示的化合物的理化性质,如下。
(1)外观:无色油状物
(2)分子式:C21H29O3N
(3)高分辨质谱分析(HRESI-MS)(m/z):
实验值344.2222(M+H)+
计算值344.2220(以C21H30O3N计)
(4)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:2953,2922,1743,1635,1617,1558,1507,1214,994,760,688
(5)质子核磁共振光谱(400MHz,CDCl3):
δ=0.89(3H,t,J=6.6),1.21-1.51(10H,m),1.60(2H,m),2.22(3H,s),2.74(2H,br),3.80(3H,s),4.90(2H,s),7.20(1H,d,J=8.7),7.33(1H,ddd,J=8.0,6.8,0.7),7.58(1H,ddd,J=8.7,7.1,1.6),8.47(1H,dd,J=8.0,1.6)
(6)碳13核磁共振光谱(100MHz,CDCl3):
δ=11.65,14.06,22.60,28.06,29.13,29.17,29.75,30.93,31.75,48.47,53.01,114.19,117.55,123.11,124.79,127.30,131.89,140.66,150.66,168.69,177.39
(制造例5)
<结构式(6)表示的化合物的制造>
按以下方式,通过化学合成制造所述结构式(6)表示的化合物。
-结构式(6)表示的化合物的制造-
使所述结构式(5)表示的化合物(890mg,2.59mmol)溶解在乙醇(以下有时称为“EtOH”。5mL)与THF(5mL)的混合溶剂中,加入2M氢氧化钠水溶液(2.0mL),在室温下使其搅拌2小时。冰浴下加入1N盐酸将pH调到4,用乙酸乙酯萃取。对有机层进行芒硝干燥,使溶剂蒸馏除去,从而获得结构式(6)表示的化合物(530mg,62%)。
--理化性质--
作为所述结构式(6)表示的化合物的理化性质,如下。
(1)外观:白色粉状
(2)熔点:161℃~163℃
(3)分子式:C20H27O3N
(4)高分辨质谱分析(HRESI-MS)(m/z):
实验值330.2063(M+H)+
计算值330.2064(以C20H28O3N计)
(5)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:2955,2925,2853,1725,1635,1593,1506,1191,976,760,689
(6)质子核磁共振光谱(400MHz,CDCl3):
δ=0.87(3H,t,J=6.4),1.20-1.65(12H,m),2.19(3H,s),2.80(2H,br),4.98(2H,br),7.26(1H,t,J=8.0),7.42(1H,d,J=8.7),7.54(1H,ddd,J=8.7,6.8,1.1),8.39(1H,dd,J=8.0,1.1)
(7)碳13核磁共振光谱(100MHz,CDCl3):
δ=11.91,14.06,22.68,27.85,29.12,29.78,31.26,31.73,49.71,115.46,117.21,123.86,123.94,126.69,132.47,140.53,154.22,169.35.176.57
(制造例6)
<结构式(1)表示的化合物的制造>
按以下方式,通过化学合成制造所述结构式(1)表示的化合物。
-结构式(1)表示的化合物的制造-
使所述结构式(21)(100mg,0.37mmol)溶解在N,N-二甲基甲酰胺(以下有时称为“DMF”。5mL),加入碳酸钾(332mg,2.40mmol)、溴乙酸甲酯(53.0mL,0.56mmol),在80℃下使其搅拌12小时。加入水,使反应停止,用乙酸乙酯萃取。对有机层进行芒硝干燥,蒸馏除去溶剂。将得到的残渣采用硅胶色层分离法(己烷∶乙酸乙酯=2∶1)纯化,获得结构式(1)表示的化合物(83.0mg,66%)。
--理化性质--
作为所述结构式(1)表示的化合物的理化性质,如下。
(1)外观:无色油状物
(2)分子式:C21H29O3N
(3)高分辨质谱分析(HRESI-MS)(m/z):
实验值344.2221(M+H)+
计算值344.2220(以C21H30O3N计)
(4)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:2953,2925,2854,1765,1618,1596,1437,1123,968,768,680
(5)质子核磁共振光谱(400MHz,CDCl3):
δ=0.87(3H,t,J=6.6),1.20-1.48(10H,m),1.71(2H,m),2.43(3H,s),2.95(2H,m),3.86(3H,s),4.62(2H,s),7.47(1H,ddd,J=8.2,6.8,1.1),7.62(1H,ddd,J=8.4,6.8,1.3),8.00(1H,d,J=8.4),8.05(1H,d,J=8.2)
(6)碳13核磁共振光谱(100MHz,CDCl3):
δ=11.90,14.08,22.63,28.99,29.23,29.49,29.86,31.83,37.02,52.32,70.26,120.85,120.73,121.39,121.76,125.73,128.82,128.85,147.84,159.03,164.32,168.95
(制造例7)
<结构式(2)表示的化合物的制造>
按以下方式,通过化学合成制造所述结构式(2)表示的化合物。
-结构式(2)表示的化合物的制造-
使所述结构式(1)表示的化合物(80.0mg,0.233mmol)溶解在EtOH(1mL)与THF(1mL)的混合溶剂中,加入2M氢氧化钠水溶液(0.5mL),在室温下使其搅拌2小时。冰浴下加入1N盐酸将pH调成4,用乙酸乙酯萃取。对有机层进行芒硝干燥,使溶剂蒸馏除去,获得结构式(2)表示的化合物(68.2mg,88%)。
--理化性质--
作为所述结构式(2)表示的化合物的理化性质,如下。
(1)外观:白色粉状
(2)熔点:59℃~62℃
(3)分子式:C20H27O3N
(4)高分辨质谱分析(HRESI-MS)(m/z):
实验值330.2064(M+H)+
计算值330.2064(以C20H28O3N计)
(5)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:2927,2855,2713,1736,1642,1589,1227,1181,1078,764,724
(6)质子核磁共振光谱(600MHz,Methanol-d4):
δ=0.88(3H,t,J=6.8),1.25-1.41(8H,m),1.50(2H,m),1.78(2H,m),2.54(3H,s),3.15(2H,t,m),4.92(2H,s),7.54(1H,ddd,J=8.2,7.2,1.0),7.92(1H,ddd,J=8.5,6.8,1.0),8.07(1H,brd,J=8.5),8.42(1H,brd,J=8.2)
(7)碳13核磁共振光谱(150MHz,Methanol-d4):
δ=12.23,14.40,23.67,29.93,30.27,30.34,30.74,32.97,35.07,72.72,122.81,123.71,123.85,124.62,129.16,133.85,142.14,164.30,167.46,171.91
(制造例8)
<结构式(7)表示的化合物的制造>
按以下方式,通过化学合成制造所述结构式(7)表示的化合物。
-结构式(7)表示的化合物的制造-
使所述结构式(21)表示的化合物(50.0mg,0.184mmol)溶解在THF(1.0mL)中,加入叔丁醇锂(29.4mg,0.37mmol),在室温下使其搅拌20分钟。接着,加入溴化氰(0.6mL,1.84mmol),在室温下使其搅拌2小时。加入水,使反应停止,用乙酸乙酯萃取。对有机层进行芒硝干燥,蒸馏除去溶剂。将得到的残渣采用硅胶色层分离法(己烷∶乙酸乙酯=2∶1)纯化,获得结构式(7)表示的化合物(34.0mg,62%)。
--理化性质--
作为所述结构式(7)表示的化合物的理化性质,如下。
(1)外观:无色油状物
(2)分子式:C19H24ON2
(3)高分辨质谱分析(HRESI-MS)(m/z):
实验值297.1961(M+H)+
计算值297.1961(以C19H25ON2计)
(4)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:2961,2926,28532237,1628,1576,1470,1292,1191,761,693
(5)质子核磁共振光谱(400MHz,CDCl3):
δ=0.87(3H,t,J=6.8),1.20-1.50(10H,m),1.71(2H,m),2.15(3H,s),2.93(2H,m),7.47(1H,m),7.73(2H,m),8.33(1H,ddd,J=8.0,0.92,1.1)
(6)碳13核磁共振光谱(100MHz,CDCl3):
δ=11.20,14.05,22.59,28.00,29.07,29.11,29.42,31.73,31.80,106.42,116.28,120.30,123.35,126.23,127.08,133.34,137.25,146.10.177.31
(制造例9)
<结构式(9)表示的化合物的制造>
按以下方式,通过化学合成制造所述结构式(9)表示的化合物。
-结构式(9)表示的化合物的制造-
使所述结构式(6)表示的化合物(100mg,0.30mmol)悬浮在THF(5mL)中,加入三乙基胺(50.8mL,0.36mmol)、叠氮磷酸二苯酯(以下有时称为“DPPA”。72.0mL,0.33mmol)、甲硫醇钠(23.0mg,0.334mmol),回流下搅拌2小时。加入氯化铵水溶液,用乙酸乙酯萃取。对有机层进行芒硝干燥,蒸馏除去溶剂。将得到的残渣采用硅胶色层分离法(己烷∶乙酸乙酯=3∶1)纯化,获得结构式(9)表示的化合物(39.3mg,36%)。
--理化性质--
作为所述结构式(9)表示的化合物的理化性质,如下。
(1)外观:无色油状物
(2)分子式:C21H29O2NS
(3)高分辨质谱分析(HRESI-MS)(m/z):
实验值360.1994(M+H)+
计算值360.1992(以C21H30O2NS计)
(4)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:2924,2852,1687,1614,1594,1542,1193,1028,757,558
(5)质子核磁共振光谱(400MHz,CDCl3):
δ=0.88(3H,t,J=6.4),1.20-1.50(10H,m),1.60(2H,br),2.23(3H,s),2.33(3H,s),2.51-2.99(2H,br),5.01(2H,br),7.21(1H,d,J=8.7),7.34(1H,dd,J=8.0、6.6),7.58(1H,ddd,J=8.7,6.6,1.4),8.47(1H,dd,J=8.0,1.4)
(6)碳13核磁共振光谱(100MHz,CDCl3):
δ=11.38,11.68,14.06,22.60,28.16,29.13,29.16,29.75,31.03,31.74,55.95,114.61,117.94,123.32,127.29,131.97,132.02,140.73,150.65,177.49,196.82
(制造例10)
<结构式(10)表示的化合物的制造>
按以下方式,通过化学合成制造所述结构式(10)表示的化合物。
-结构式(10)表示的化合物的制造-
使所述结构式(6)表示的化合物(100mg,0.30mmol)悬浮在THF(5mL)中,在冰冷却下加入三乙基胺(50.8mL,0.365mmol)、DPPA(72.0mL,0.33mmol),进一步回流下搅拌1小时。在其中加入甲硫醇钠(23.0mg,0.33mmol),进一步搅拌1小时。加入氯化铵水溶液,用乙酸乙酯萃取。对有机层进行芒硝干燥,蒸馏除去溶剂。将得到的残渣采用硅胶色层分离法(己烷∶乙酸乙酯=5∶1)纯化,获得结构式(10)表示的化合物(48.6mg,43%)。
--理化性质--
作为所述结构式(10)表示的化合物的理化性质,如下。
(1)外观:无色油状物
(2)分子式:C21H30O2N2S
(3)高分辨质谱分析(HRESI-MS)(m/z):
实验值397.1921(M+Na)+
计算值397.1920(以C21H30O2N2NaS计)
(4)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:3169,2955,2927,1671,1615,1595,1556,1492,1195,1084,760,651
(5)质子核磁共振光谱(600MHz,CDCl3):
δ=0.89(3H,t,J=6.7),1.22-1.45(15H,m),2.45(2H,br),2.46(3H,s),5.67(2H,br),7.24(1H,ddd,J=7.9,6.8,1.0),7.49(1H,d,J=8.6),7.59(1H,ddd,J=8.6,6.8,1.4),8.26(1H,dd,J=7.9,1.4),8.78(1H,br)
(6)碳13核磁共振光谱(150MHz,CDCl3):
δ=11.05,12.22,14.02,22.59,28.48,29.11,29.16,29.73,30.73,31.77,52.58,115.46,117.00,123.24,124.37,126.89,132.32,139.56,151.60,168.61,177.27
(制造例11)
<结构式(11)表示的化合物的制造>
按以下方式,通过化学合成制造所述结构式(11)表示的化合物。
-结构式(15)表示的化合物的制造-
氩气氛下使所述结构式(14)表示的化合物(1当量)溶解在二氯甲烷中,加入三乙基胺(2当量)。进一步在冰浴下滴加作为酰氯的辛酰氯(3.0当量),在室温下使其搅拌。用0.1N盐酸停止反应,用二氯甲烷萃取,接着,用饱和碳酸氢钠水溶液、食盐水洗涤。合并有机层进行芒硝干燥,蒸馏除去溶剂。能够采用硅胶色层分离法(己烷∶乙酸乙酯)纯化得到的残渣而获得结构式(15)表示的化合物。
--理化性质--
作为所述结构式(15)表示的化合物的理化性质,如下。
(1)外观:无色油状物
(2)分子式:C17H25O2N
(3)高分辨质谱分析(HRESI-MS)(m/z):
实验值298.1777(M+Na)+
计算值298.1778(以C17H25O2NNa计)
(4)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:3255,2954,2928,1698,754
(5)质子核磁共振光谱(400MHz,CDCl3):
δ=0.87(3H,t,J=6.6),1.20-1.41(11H,m),1.74(2H,m),2.43(2H,t,J=7.5),3.08(2H,q,J=7.1),7.09(1H,ddd,J=8.2,7.1,1.4),7.53(1H,ddd,J=8.4,7.1,1.4),7.92(1H,dd,J=8.2,1.4),8.77(1H,dd,J=8.4,1.4),11.65(1H,br)
(6)碳13核磁共振光谱(100MHz,CDCl3):
δ=8.44,14.05,22.59,25.53,28.98,29.16,31.67,33.13,38.78,120.83,121.37,122.11,130.56,134.82,141.06,172.78,205.36
-结构式(20)表示的化合物的制造-
在所述结构式(15)表示的化合物(1当量)的二噁烷0.1M溶液中加入氢氧化钠(3.0当量),在110℃下使其搅拌1小时~2小时。将反应溶液恢复到室温,加入水,进一步加入1N盐酸直至pH变成7。进一步加入己烷施加超声波,则固体析出。抽滤固体,用水、己烷洗涤。能够洗涤后使其干燥,从而获得结构式(20)表示的化合物。
--理化性质--
作为所述结构式(20)表示的化合物的理化性质,如下。
(1)外观:白色粉状
(2)熔点:228℃~231℃
(3)分子式:C17H23ON
(4)高分辨质谱分析(HRESI-MS)(m/z):
实验值258.1853(M+H)+
计算值258.1852(以C17H24ON计)
(5)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:3061,2954,2925,1637,1608,1556,1504,1188,998,754,692
(6)质子核磁共振光谱(400MHz,Methanol-d4):
δ=0.88(3H,t,J=6.6),1.30-1.46(8H,m),1.65-1.73(2H,m),2.14(3H,s),2.78(2H,t,J=7.7),7.32(1H,t,J=8.1),7.52(1H,d,m),7.61(1H,m),8.21(1H,d,J=8.2)
(7)碳13核磁共振光谱(100MHz,Methanol-d4):
δ=10.84,14.40,23.68,29.99,30.16,30.54,32.91,33.45,116.16,118.67,124.38,124.51,126.15,132.65,140.55,153.37,179.53
-结构式(11)表示的化合物的制造-
氩气氛下使所述结构式(20)表示的化合物(1.0g,3.89mmol)溶解在THF(10mL)中,加入叔丁醇锂的THF溶液(5.8mL,5.80mmol),在室温下使其搅拌20分钟。冰冷却下在其中滴加硫氰酸氯甲酯(6.7mL,38.9mmol),进一步在室温下使其搅拌2小时。加入食盐水,使反应停止,用乙酸乙酯萃取。对有机层进行芒硝干燥后,将得到的残渣采用硅胶色层分离法(己烷∶乙酸乙酯=2∶1)纯化,获得结构式(11)表示的化合物(330mg,26%)。
--理化性质--
作为所述结构式(11)表示的化合物的理化性质,如下。
(1)外观:黄色油状物
(2)分子式:C19H24ON2S
(3)高分辨质谱分析(HRESI-MS)(m/z):
实验值329.1682(M+H)+
计算值329.1682(以C19H25ON2S计)
(4)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:2961,2926,2853,2237,1628,1576,1470,1292,1191,987,761,693
(5)质子核磁共振光谱(400MHz,CDCl3):
δ=0.90(3H,t,J=6.6),1.23-1.71(10H,m),2.19(3H,s),2.84(2H,m),5.71(2H,s),7.38(1H,ddd,J=8.0,6.8,0.9),7.46(1H,d,J=8.7),7.68(1H,ddd,J=8.7,6.8,1.6),8.45(1H,dd,J=8.0,1.6)
(6)碳13核磁共振光谱(100MHz,CDCl3):
δ=11.60,14.05,22.56,28.59,28.88,29.76,30.55,31.66,56.26,114.42,118.19,123.83,124.60,127.31,132.41,139.86,141.68,149.60,177.64
(制造例12)
<结构式(12)表示的化合物的制造>
按以下方式,通过化学合成制造所述结构式(12)表示的化合物。
-结构式(12)表示的化合物的制造-
在所述结构式(11)表示的化合物(100mg,0.30mmol)与甲硫醇钠(23.4mg,0.33mmol)的混合物中加入乙腈(1.5mL),在室温下搅拌20分钟。加入饱和碳酸氢钠水溶液,用乙酸乙酯萃取。对有机层进行芒硝干燥后,将得到的残渣采用硅胶色层分离法(己烷∶乙酸乙酯=2∶1)纯化,获得结构式(12)表示的化合物(31.7mg,27%)。
--理化性质--
作为所述结构式(12)表示的化合物的理化性质,如下。
(1)外观:黄色粉状
(2)熔点:167℃~170℃;
(3)分子式:C20H28ON2S2
(4)高分辨质谱分析(HRESI-MS)(m/z):
实验值399.1534(M+Na)+
计算值399.1535(以C20H28ON2NaS2计)
(5)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:3119,2958,2918,2850,1619,1598,1538,1282,1199,1105,938,764,688
(6)质子核磁共振光谱(400MHz,CDCl3):
δ=0.86(3H,t,J=6.6),1.21-1.50(13H,m),2.22-2.58(2H,br),2.76(3H,s),5.68-6.41(2H,br),7.22(1H,t,J=7.8),7.44(1H,d,J=8.7),7.59(1H,ddd,J=8.7,7.8,1.1),8.15(1H,d,J=7.8),10.09(1H,br)
(7)碳13核磁共振光谱(100MHz,CDCl3):
δ=10.74,14.02,18.01,22.53,28.29,28.77,29.64,30.85,31.59,58.21,115.93,116.84,123.54,123.91,126.35,132.65,139.39,152.23,177.13,199.84
(制造例13)
<结构式(13)表示的化合物的制造>
按以下方式,通过化学合成制造所述结构式(13)表示的化合物。
-结构式(13)表示的化合物的制造-
在所述结构式(11)表示的化合物(50.0mg,0.15mmol)与甲硫醇钠(10.6mg,0.15mmol)的混合物中加入乙腈(1.5mL),在室温下搅拌20分钟。接着,在室温下加入碘甲烷(8.5mL,0.17mmol),进一步搅拌30分钟。加入饱和碳酸氢钠水溶液,用乙酸乙酯萃取。对有机层进行芒硝干燥后,将得到的残渣采用硅胶色层分离法(己烷∶乙酸乙酯=2∶1)纯化,获得结构式(13)表示的化合物(30.6mg,51%)。
--理化性质--
作为所述结构式(13)表示的化合物的理化性质,如下。
(1)外观:白色粉状
(2)熔点:92℃~94℃
(3)分子式:C21H30ON252
(4)高分辨质谱分析(HRESI-MS)(m/z):
实验值413.1689(M+Na)+
计算值413.1692(以C21H30ON2NaS2计)
(5)红外线吸收光谱:
经KBr片剂法测定的红外线吸收的峰如下。
νmax(KBr)cm-1:2958,2922,2852,1618,1595,1566,1492,1370,1277,1192,1004,769,700
(6)质子核磁共振光谱(400MHz,CDCl3).
δ=0.89(3H,t,J=6.8),1.24-1.50(8H,m),1.64(2H,m),2.22(3H,s),2.28(3H,s),2.71(3H,s),2.77(2H,m),5.60(2H,s),7.31(2H,m),7.55(1H,ddd,J=8.4,7.1,1.6),8.46(1H,dd,J=8.0,1.6)
(7)碳13核磁共振光谱(100MHz,CDCl3):
δ=11.50,14.06,14.75,15.03,22.61,28.28,28.92,29.83,30.66,31.76,63.75,115.84,116.97,122.77,124.81,126.75,131.27,141.06,151.35,161.39,177.54
(试验例1:体外(In vitro)的抗癌活性)
将所述制造例1~13中得到的结构式(1)~(13)表示的化合物的In vitro的抗癌活性按以下方式进行试验。
<细胞增殖试验1(人胃癌细胞MKN-74)>
-细胞的制备-
将人胃癌细胞MKN-74(理研Cell Bank)在添加有10%FBS(GIBCO公司制)、100units/mL的青霉素G(Invitrogen公司制)、100μg/mL的链霉素(Invitrogen公司制)的DMEM中在37℃、5%CO2下培养。
利用Lipofectamine试剂(Invitrogen公司制)对所述癌细胞基因转入绿色荧光蛋白(Green fluorescence protein,GFP)表达载体pEGFP-C1(BD Biosciences公司制),将稳定地表达GFP的细胞克隆。
将来自人胃的正常间质细胞Hs738((CRL-7869),ATCC)在添加有10%FBS、100units/mL的青霉素G(Invitrogen公司制)、100μg/mL的链霉素(Invitrogen公司制)、5μg/mL的胰岛素、5μg/mL的转铁蛋白(和光纯药工业公司制)、1.4μM的氢化可的松(Sigma公司制)、5mg/mL的basic-FGF(Pepro Tech公司制)的DMEM中在37℃、5%CO2下培养。
-共培养试验-
使所述来自人胃的正常间质细胞Hs738在含有1%透析血清、5μg/mL的胰岛素、5μg/mL的转铁蛋白(和光纯药工业公司制)、1.4μM的氢化可的松(Sigma公司制)的DMEM中以5×104个/mL分散,以0.1mL/孔铺撒在96孔板中,在各浓度下加入各评价样品(结构式(1)~(13)表示的化合物),在37℃、5%CO2下培养2天。
接着,使所述人胃癌细胞MKN-74在DMEM中以5×105个/mL分散,以10μL/孔铺撒在培养了所述来自人胃的正常间质细胞Hs738的板中,进一步在37℃、5%CO2下共培养3天。
-单独培养试验-
仅将含有1%透析血清、5μg/mL的胰岛素、5μg/mL的转铁蛋白(和光纯药工业公司制)、1.4μM的氢化可的松(Sigma公司制)的DMEM以0.1mL/孔铺撒在96孔板中,在各浓度下加入各评价样品(结构式(1)~(13)表示的化合物),在37℃、5%CO2下保持2天。
接着,使所述人胃癌细胞MKN-74在DMEM中以5×105个/mL分散,以10μL/孔铺撒在所述板中,进一步在37℃、5%CO2下共培养3天。
-细胞增殖率的测定-
所述共培养试验和单独培养试验中细胞增殖率的测定按以下方式进行。
从所述板的孔中除去培养基,以0.1mL/孔加入细胞裂解液(10mM Tris-HCl[pH 7.4]、150mM NaCl、0.9mM CaCl2、1%Triton X-100),裂解细胞,在激发波长为485nm、荧光波长为538nm下测定GFP的荧光强度,由下述式计算出细胞增殖率。将由所述细胞增殖率的结果计算出IC50的结果示于表1。
细胞增殖率(%)=(有评价样品的荧光强度/没有评价样品的荧光强度)×100
[表1]
表1中的值表示2系列的平均值,标准误差(SE)为10%以下。
表1中,“Mono”表示单独培养试验的结果,“Cocul”表示共培养试验的结果。
如表1所示,对于所述结构式(1)~(13)表示的13种化合物,均与仅培养胃癌细胞MKN-74的(Mono)进行比较,以更低浓度的IC50值强烈地阻碍与胃间质细胞共培养时(Cocul)的胃癌细胞MKN-74的增殖。
(试验例2:体内(In vivo)的抗癌活性)
将所述制造例中得到的结构式(2)表示的化合物、和结构式(13)表示的化合物的In vivo的抗癌活性按以下方式试验。
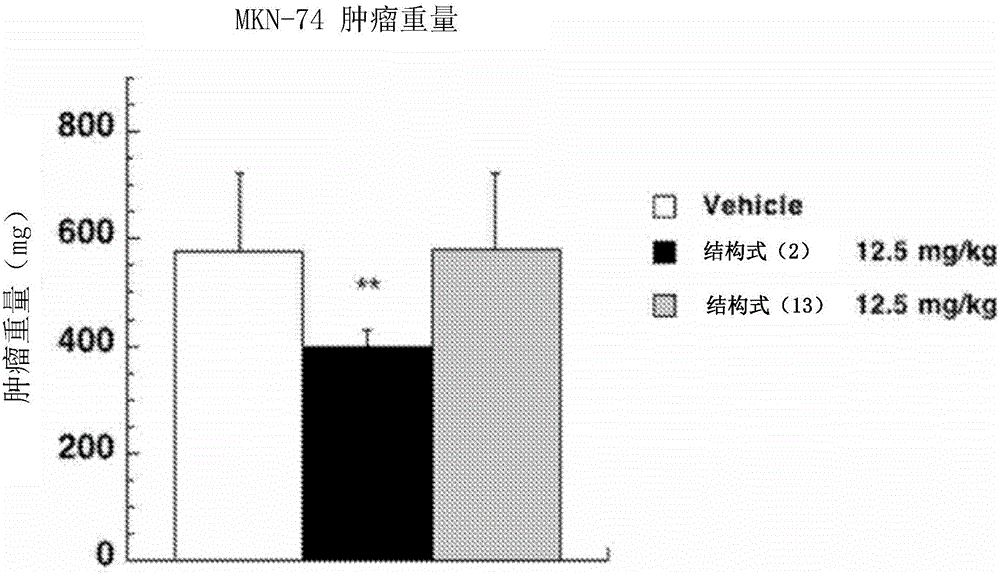
<试验例2-1:单独人胃癌细胞MKN-74>
将BALB/c nu/nu裸鼠(雌,5周龄,Charles River公司制)在SPF条件下饲养。
对培养的人胃癌细胞MKN-74进行胰蛋白酶处理,将从培养皿中剥落的所述人胃癌细胞MKN-74(8×106个)分散在含有0.3mL的10%FBS的DMEM中,与0.5mL的减少生长因子基底膜(growth factor-reduced Matrigel,BD Biosciences公司制)混合。
将所述混合的细胞液0.1mL(癌细胞1×106个)皮下接种在所述小鼠的左鼠胫部中。
将所述结构式(2)表示的化合物、或结构式(13)表示的化合物在规定的时间向静脉内给药,切下皮下出现的肿瘤,测定其重量。应予说明,所述结构式(2)表示的化合物、或结构式(13)表示的化合物的给药量以每个给药日的量计为12.5mg/kg。
另外,肿瘤体积参照所述非专利文献1,由以下式计算出。
肿瘤体积(mm3)=(长径×短径2)/2
应予说明,对给予了生理食盐水来代替结构式(2)表示的化合物、或结构式(13)表示的化合物的(vehicle)也同样地进行试验作为对照。
<试验例2-2:人胃癌细胞MKN-74和来自人胃的正常间质细胞Hs738>
在试验例2-1中,使单独使用人胃癌细胞MKN-74这点为人胃癌细胞MKN-74和来自人胃的正常间质细胞Hs738,除此之外,与试验例2-1同样地进行试验。
应予说明,细胞液和对小鼠接种细胞液按以下方式进行。
分别对培养的人胃癌细胞MKN-74和来自人胃的正常间质细胞Hs738进行胰蛋白酶处理,从培养皿中剥落。将所述人胃癌细胞MKN-74(8×106个)、和所述来自人胃的正常间质细胞Hs738(8×106个)分散在含有0.3mL的10%FBS的DMEM中,与0.5mL的减少生长因子基底膜(growth factor-reduced Matrigel,BD Biosciences公司制)混合。
将所述混合的细胞液0.1mL(癌细胞1×106个、和间质细胞1×106个的混合)皮下接种在所述小鼠的左鼠胫部。
将所述试验例2的结果示于图1A~图1D。
图1A表示所述试验例2-1中的肿瘤体积的变化,图1B表示所述试验例2-1中的肿瘤重量(从肿瘤接种起第21天),图1C表示所述试验例2-2中的肿瘤体积的变化,图1D表示所述试验例2-2中的肿瘤重量(从肿瘤接种起第21天)。
图1A和图1C中,“○”表示“vehicle”的结果,“●”表示“结构式(2)表示的化合物的给药量为12.5mg/kg”的结果,“□”表示“结构式(13)表示的化合物的给药量为12.5mg/kg”的结果。另外,图1A和图1C中,“箭头”表示将结构式(2)表示的化合物、或结构式(13)表示的化合物给药的天数。
图1B和图1D中,“白”表示“vehicle”的结果,“黑”表示“结构式(2)表示的化合物的给药量为12.5mg/kg”的结果,“灰”表示“结构式(13)表示的化合物的给药量为12.5mg/kg”的结果。
图1A~图1D中的值表示5只小鼠的平均值与标准偏差(SD),*表示P<0.05,**表示P<0.01。
如图1A~图1D所示,对于单独人胃癌细胞MKN-74来说,通过将结构式(2)表示的化合物以12.5mg/kg静脉内给药而有意义地抑制。
另一方面,对于与来自人胃的间质细胞Hs738一起移植的肿瘤来说,通过将结构式(13)表示的化合物以12.5mg/kg静脉内给药而有意义抑制。
(试验例3:急性毒性试验)
将ICR小鼠(雌,4周龄,Charles River公司制)在SPF条件下饲养。
作为评价样品,将所述结构式(1)~(13)表示的化合物进行静脉内给药并观察小鼠2周。2周的观察期间中,以确认了死亡或重度的毒性的给药量的二分之一量作为本实验中的最大耐容量(MTD)。将结果示于表2。
[表2]
由所述表2的结果可知,将所述结构式(1)表示的化合物、结构式(2)表示的化合物、结构式(6)表示的化合物、结构式(9)表示的化合物、结构式(10)表示的化合物、结构式(12)表示的化合物、结构式(13)表示的化合物进行静脉内给药的情况下的MTD为50mg/kg以上。
(试验例4:抗菌活性)
将所述制造例中得到的结构式(2)表示的化合物、结构式(3)表示的化合物、结构式(4)表示的化合物、结构式(5)表示的化合物、结构式(6)表示的化合物、结构式(8)表示的化合物、结构式(11)表示的化合物、结构式(12)表示的化合物、和结构式(13)表示的化合物的抗菌活性按以下方式进行试验。
应予说明,作为比较,对于克拉霉素(Clarithromycin)、和氨苄青霉素(ABPC)也同样地进行试验。
<试验例4-1:针对幽门螺杆菌测定MIC>
进行所述各化合物针对幽门螺杆菌的最小抑菌浓度(MIC)的测定。
将幽门螺杆菌(Helicobacter pylori)JCM12093株、和幽门螺杆菌(H.pylori)JCM12095株在HP培养基(在脑心浸液肉汤培养基(Becton Dickinson公司制)中添加10%胎牛血清(Life Technologies公司制))中在37℃、微需氧培养条件下(微需氧条件(N2:O2:CO2=85:5:10))静置培养144小时。培养完成后,将培养液在HP培养基中悬浮,以幽门螺杆菌为2×106CFU/mL~9×106CFU/mL的方式稀释。
各试验样品(所述各化合物、克拉霉素、氨苄青霉素)分别在HP培养基中以256mg/L制备。由此,进行两倍阶段稀释,进行15阶段的稀释到0.0078mg/L为止。
分别在含有所述各浓度的试验样品的50μL/孔的HP培养基中以50μL/孔添加所述稀释了的各菌液,在37℃、微需氧培养条件下(微需氧条件(N2:O2:CO2=85:5:10))下静置培养144小时。
培养完成后,通过浊度目视判断各菌是否增殖,求得各菌株的MIC。将结果示于表3。
<试验例4-2:针对黄色葡萄球菌和大肠杆菌测定MIC>
进行所述各化合物针对黄色葡萄球菌和大肠杆菌的最小抑菌浓度(MIC)的测定。
将金黄色葡萄球菌(Staphylococcus aureus)FDA209P株、和大肠杆菌(Escherichia coli)K-12株在普通肉汤培养基(Polypeptone(日本制药公司制)1%、细菌用鱼提取物(极东制药工业公司制)1%、氯化钠0.2%)中在37℃下振荡培养一夜。培养完成后在普通肉汤培养基中以各细菌为2×106CFU/mL~9×106CFU/mL的方式稀释。
各试验样品(所述各化合物、克拉霉素、氨苄青霉素)分别在普通肉汤培养基中以256mg/L制备。由此,进行两倍阶段稀释,进行11阶段的稀释到0.125mg/L为止。
分别在含有所述各浓度的试验样品的50μL/孔的普通肉汤培养基中以50μL/孔添加所述稀释了的各菌液,在37℃下静置培养一夜。
培养完成后,通过浊度目视判断各菌是否增殖,求得各菌株的MIC。将结果示于表3。
<试验例4-3:针对肠球菌测定MIC>
进行所述各化合物针对肠球菌的最小抑菌浓度(MIC)的测定。
将粪肠球菌(Enterococcus faecalis)JCM5803株在心浸汤培养基(Becton Dickinson公司制)中在37℃下振荡培养一夜。培养完成后,用心浸汤培养基稀释,以各细菌为2×104CFU/mL~9×104CFU/mL的方式稀释。
各试验样品(所述各化合物、克拉霉素、氨苄青霉素)分别在心浸汤培养基中以256mg/L制备。由此,进行两倍阶段稀释,进行11阶段的稀释到0.125mg/L为止。
分别在含有所述各浓度的试验样品的50μL/孔的心浸汤培养基中以50μL/孔添加所述稀释了的各菌液,在37℃下静置培养18小时。
培养完成后,通过浊度目视判断菌是否增殖,求得菌株的MIC。将结果示于3。
<试验例4-4:针对流感嗜血杆菌测定MIC>
进行所述各化合物针对流感嗜血杆菌的最小抑菌浓度(MIC)的测定。
将流感嗜血杆菌(Haemophilus influenzae)T-196株、和流感嗜血杆菌(H.influenzae)ARD476株在HI培养基(在Muller Hinton培养基(Becton Dickinson公司制)中添加5%的Fildes强化营养(Fildes enrichment,Becton Dickinson公司制))中、在37℃、含5%碳酸气的需氧条件下静置培养一夜。
培养完成后,将培养液在HI培养基中悬浮,以各流感嗜血杆菌为2×106CFU/mL~9×106CFU/mL的方式稀释。
各试验样品(所述各化合物、克拉霉素、氨苄青霉素)分别在HI培养基中以256mg/L制备。由此,进行两倍阶段稀释,进行11阶段稀释到0.125mg/L为止。
分别在含有所述各浓度的试验样品的50μL/孔的HI培养基中以50μL/孔添加所述稀释了的各菌液,在37℃、含5%碳酸气的需氧条件下静置培养18小时。
培养完成后,通过浊度目视判断菌是否增殖,求得菌株的MIC。将结果示于3。
[表3]
如表3所示,所述各化合物显示出抗幽门螺杆菌活性。在这些之中,结构式(3)表示的化合物、结构式(11)表示的化合物、结构式(12)表示的化合物均以低浓度显示出抗幽门螺杆菌活性。
另一方面,所述各化合物针对其它通常的感染病原菌为弱的抗菌活性。
本发明的任一个结构式(1)~(13)表示的化合物具有优异的抗癌作用或优异的抗幽门螺杆菌活性,是安全性高的化合物,因此,可以优选用作药品组合物、抗癌剂、抗幽门螺杆菌剂等有效成分。
- 还没有人留言评论。精彩留言会获得点赞!