一种减少扩增偏倚的基因突变检测方法和试剂与流程
本发明涉及基因检测
技术领域:
,尤其涉及一种减少扩增偏倚的基因突变检测方法和试剂。
背景技术:
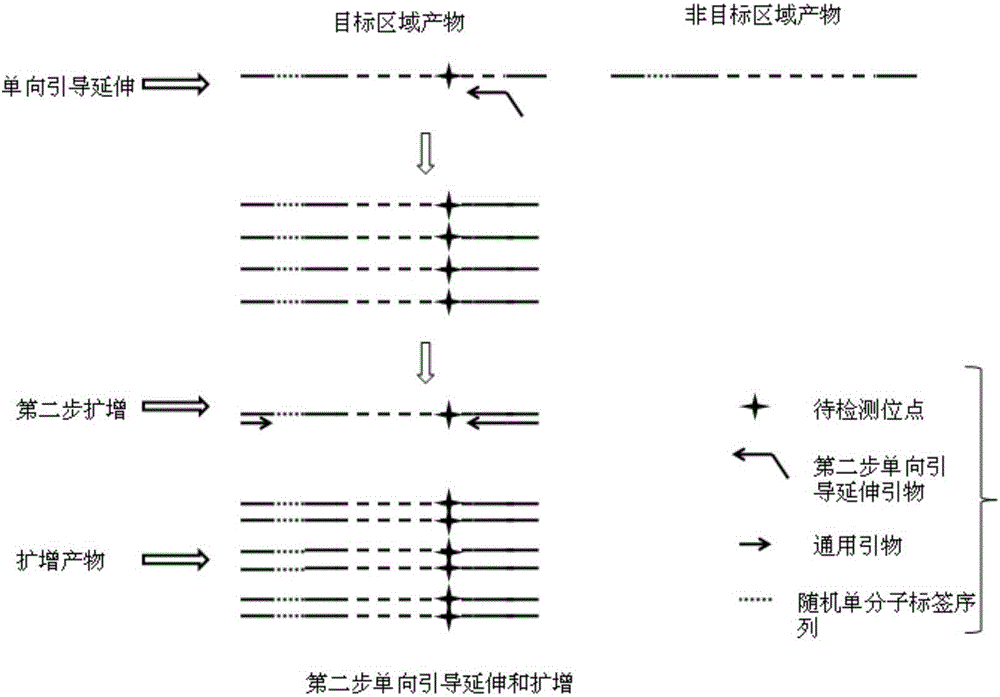
:基因检测是通过血液、其他体液或细胞对受检者的DNA进行检测的技术。基因检测可以诊断疾病,也可以用于疾病风险的预测。通过基因检测进行疾病早期筛查、疾病诊断、伴随治疗时常常需要对多个低丰度基因突变位点进行准确灵敏的检测。例如,肿瘤是高度异质性的,其中致病突变可能以极低比例存在。实现肿瘤的精准基因检测需要从大量的非肿瘤DNA和非致病突变的肿瘤DNA中找到相关的低丰度基因突变。目前常用的基因检测方法均以PCR(聚合酶链式反应)为基础。PCR扩增为指数扩增,即在扩增的指数期会使PCR产物达到2n倍(n为进入指数期的PCR循环数)。因此PCR扩增会产生扩增偏倚,即原本丰度较高的DNA分子在扩增产物中所占的比例会更大,原有的低丰度DNA分子在扩增产物中所占的比例变得更小。这样就容易出现低丰度DNA分子在PCR产物中被湮没的情况,最终使得检测灵敏度变低,甚至出现某些检测位点假阴性(即无法检测出)的情况。在高通量测序中,PCR扩增偏倚也会导致大量的冗余数据产生,大大增加了测序成本。DSN(Duplex-SpecificNuclease)是一种来自红金蟹的双链特异性核酸酶,具有热稳定性,在60-65℃时具有最大活力。DSN能够高选择性的识别并酶切完全匹配的双链DNA,对单链DNA几乎无作用。在高通量测序平台文库构建的某个时段适当使用DSN核酸酶进行处理可以有效减少PCR扩增产生的偏倚效应。目前现有的减少PCR扩增偏倚的技术解决方案主要是从控制PCR指数期的角度进行的。例如在文库构建过程中尽量减少PCR循环数,未达到指数期或指数期扩增越少产生的扩增偏倚程度就越小。但是减少PCR循环数将直接导致扩增产物得率低,尤其当待检测DNA材料量极少时无法产生足够的PCR产物用于检测,并且低丰度DNA突变很难达到足够的拷贝数以用于检测。另一方案是使用扩增均一度表现优越的DNA聚合酶,此方案只能很小程度上减少扩增偏倚,往往并不能解决实际问题。技术实现要素:本发明提供一种减少扩增偏倚的基因突变检测方法和试剂,能使高丰度DNA含量与低丰度DNA含量接近,达到减少扩增偏倚的目的,最终实现通过高通量测序技术来低成本精准检测低丰度基因突变的目的。根据本发明的第一方面,本发明提供一种减少扩增偏倚的基因突变检测方法,包括:(a)使用用于捕获待检测位点或待检测区域的第一步单向引导延伸引物对含有目标区域DNA的接头连接产物进行预定循环数的第一步单向引导延伸,然后使用第一通用引物和第一步单向引导延伸引物进行PCR扩增,其中上述第一通用引物与上述接头连接产物上的接头序列匹配;(b)使用用于捕获待检测位点或区域的第二步单向引导延伸引物对上述步骤(a)的产物进行预定循环数的第二步单向引导延伸,其中上述第二步单向引导延伸引物相比上述第一步单向引导延伸引物在模板上的结合位置距离上述待检测位点或区域更近,然后使用第二通用引物和第二步单向引导延伸引物进行PCR扩增,其中上述第二通用引物与上述接头序列匹配;(c)对上述步骤(b)的产物进行变性,然后在适于已变性的上述步骤(b)的产物退火的温度下,用双链特异性核酸酶处理上述步骤(b)的产物,以减少丰度过高的DNA分子含量,增加丰度低的DNA分子的相对含量;(d)使用第二通用引物和第三通用引物对步骤(c)的产物进行PCR扩增并测序。进一步地,上述第一步单向引导延伸引物带有用于捕获上述步骤(a)的产物的亲和标记;优选地,上述亲和标记是位于上述第一步单向引导延伸引物5’端的生物素标记。进一步地,上述第一通用引物与上述第二通用引物是相同的引物。进一步地,上述第一步单向引导延伸引物与上述第二步单向引导延伸引物均位于上述待检测位点或待检测区域在上述目标区域DNA的同方向。进一步地,上述第一步单向引导延伸引物与上述第二步单向引导延伸引物间隔距离是0-110bp,优选间隔距离是55bp。进一步地,有多个上述待检测位点或待检测区域,相应的,使用用于捕获上述多个上述待检测位点或待检测区域的多个第一步单向引导延伸引物和/或多个第二步单向引导延伸引物。进一步地,上述待检测位点或待检测区域包括点突变、插入、缺失和基因融合。进一步地,上述测序包括测序以得到上述待检测位点或待检测区域的基因突变情况。进一步地,上述方法在上述步骤(a)之前还包括:(a’)对上述接头连接产物进行预定循环数的扩增,并以扩增产物替代上述接头连接产物进行上述步骤(a);优选地,上述预定循环数是3-5个循环。根据本发明的第二方面,本发明提供一种减少扩增偏倚的基因突变检测试剂,包括:(a)用于捕获待检测位点或待检测区域的第一步单向引导延伸引物,用于对含有目标区域DNA的接头连接产物进行预定循环数的第一步单向引导延伸;和第一通用引物,用于以上述第一步单向引导延伸的产物为模板进行PCR扩增,其中上述第一通用引物与上述接头连接产物上的接头序列匹配;(b)用于捕获待检测位点或区域的第二步单向引导延伸引物,用于对上述第一通用引物的扩增产物进行预定循环数的第二步单向引导延伸,其中上述第二步单向引导延伸引物相比上述第一步单向引导延伸引物在模板上的结合位置距离上述待检测位点或区域更近;和第二通用引物,用于以上述第二步单向引导延伸的产物为模板进行PCR扩增,其中上述第二通用引物与上述接头序列匹配;(c)双链特异性核酸酶,用于在上述第二通用引物的扩增产物已变性且适于退火的温度下,处理上述扩增产物,以减少丰度过高的DNA分子含量,增加丰度低的DNA分子的相对含量;(d)第三通用引物,用于对双链特异性核酸酶处理的产物进行PCR扩增并测序。本发明的基因突变检测方法使用单向引导延伸结合DSN核酸酶处理的均一化。PCR前进行数个循环的单向引导延伸,使目标DNA的产物量呈线性增加,即每一个单向引导延伸循环会产生原模板量一倍的单向引导延伸产物。多个单向引导延伸后产生足够的待检测DNA分子,再进行指数PCR扩增,可以相应减少扩增偏倚的程度。DSN核酸酶能够选择性消除高丰度双链DNA,最终使得高丰度DNA含量与低丰度DNA含量接近,进而达到减少扩增偏倚的目的。最终实现通过高通量测序技术来低成本精准检测低丰度基因突变的目的。附图说明图1为本发明实施例的第一步单向引导延伸和扩增过程原理示意图;图2为本发明实施例的第二步单向引导延伸和扩增过程原理示意图;图3为本发明实施例的单向引导延伸及DSN核酸酶去偏倚的效果示意图;图4为本发明实施例中纯化文库进行2%琼脂糖凝胶电泳检测的结果图,其中M表示Takara100bpmarker,A表示本发明方法文库电泳结果,B表示对照实验的文库电泳结果。图5为本发明实施例中各位点与深度比较结果,其中A表示本发明方法的各位点的深度情况,B表示对照实验的各位点的深度情况。具体实施方式下面通过具体实施方式结合附图对本发明作进一步详细说明。如图1-2所示,本发明实施例的减少扩增偏倚的基因突变检测方法,包括:(a)使用用于捕获待检测位点或待检测区域的第一步单向引导延伸引物对含有目标区域DNA的接头连接产物进行预定循环数的第一步单向引导延伸,然后使用第一通用引物进行PCR扩增,其中第一通用引物与接头连接产物上的接头序列匹配;(b)使用用于捕获待检测位点或区域的第二步单向引导延伸引物对步骤(a)的产物进行预定循环数的第二步单向引导延伸,其中第二步单向引导延伸引物相比第一步单向引导延伸引物在模板上的结合位置距离待检测位点或区域更近,然后使用第二通用引物进行PCR扩增,其中第二通用引物与接头序列匹配;(c)对步骤(b)的产物进行变性,然后在适于已变性的步骤(b)的产物退火的温度下,用双链特异性核酸酶处理步骤(b)的产物,以减少丰度过高的DNA分子含量,增加丰度低的DNA分子的相对含量;(d)使用第二通用引物和第三通用引物对步骤(c)的产物进行PCR扩增并测序。其中,第三通用引物与第一通用引物和第二通用引物在相反端,也就是结合在第一通用引物和第二通用引物所结合的一端相对的另一端的接头序列上。待检测位点或待检测区域,即基因突变所在的位点或区域,可以是单个碱基位点,也可以是一段碱基序列;其中基因突变可以是点突变、插入、缺失和基因融合等。目标区域DNA,即待检测位点或待检测区域所在的DNA区域,与此相对的,如图1所示的非目标区域DNA,即不是待检测位点或待检测区域所在的DNA区域,这样的区域也可能被第一步单向引导延伸引物结合从而发生延伸,得到不希望的扩增产物。因此,第一步单向引导延伸的产物主要是目标区域DNA的扩增产物,由于单向引导延伸特异性较差,还有少量的非目标区域DNA的扩增产物。第一步单向引导延伸过程中使用的作为扩增模板的接头连接产物是片段化的DNA连接接头序列(例如测序接头序列)的产物,其中接头序列中包含随机单分子标签序列,可以对每一个DNA分子进行特异性标记。第一通用引物(图1中的通用引物)与接头序列匹配,用于在第一步单向引导延伸之后,实现PCR扩增。第一步PCR扩增之后的产物,需要纯化,再进行第二步单向引导延伸和扩增,一种有利的方式是第一步单向引导延伸引物带有用于捕获第一步PCR扩增产物的亲和标记;优选地,位于第一步单向引导延伸引物5’端的生物素标记,可以通过生物素-亲和素的作用实现扩增产物的纯化。步骤(a)中,预定循环数可以是10-30个循环,优选15-20个循环,相应的,使原模板增加15-20倍。第二步单向引导延伸和扩增使用的引物距离待检测位点或待检测区域更近,对非目标区域DNA产物有筛选去除作用,大大增加了目标区域DNA扩增的特异性。步骤(b)中,预定循环数可以是10-30个循环,优选15-20个循环,相应的,使原模板增加15-20倍。然后,再加入通用引物(第二通用引物)进行第二步PCR扩增。在本发明实施例中,第一通用引物与第二通用引物可以是相同的引物,也可以是不同的引物,优选使用相同的引物。在本发明实施例中,优选地,第一步单向引导延伸引物与第二步单向引导延伸引物均位于待检测位点或待检测区域在目标区域DNA的同方向,图1-2中显示为,与带有随机单分子标签序列的接头相对的另一个方向。在本发明实施例中,优选地,第一步单向引导延伸引物与第二步单向引导延伸引物间隔距离是0-110bp,例如2bp、5bp、10bp、30bp、50bp、55bp、80bp或100bp等,优选间隔距离是55bp。在本发明实施例中,针对多个待检测位点或待检测区域,可以使用多个第一步单向引导延伸引物,也就是以引物组的形式提供多个第一步单向引导延伸引物的混合物,相应的,也可以以引物组的形式提供多个第二步单向引导延伸引物的混合物。在本发明实施例中,对步骤(d)的产物进行测序,即可得到待检测位点或待检测区域的基因突变情况。在本发明优选的实施例中,针对起始材料很少的DNA样本可以对连接产物进行预定循环数的扩增,即“预文库扩增”,例如进行3-5个循环的扩增。在步骤(c)中,对第二步单向引导延伸和扩增产物进行去偏倚。由于此步骤之前已进行了至少两步PCR扩增,导致扩增产物不均一。进行DSN核酸酶处理,减少丰度过高的DNA分子含量,相对增加丰度低的DNA分子含量,最终使所有待检测分子的测序深度相对均一化。此方法基于核酸杂交动力学和双链DNA特异的DSN核酸酶的独特特性。变性的DNA进行复性时,丰度高的DNA先复性,就会首先被DSN核酸酶消化降解。而DSN核酸酶对尚未复性的丰度低的单链DNA无消化降解作用。图3示出了一个本发明实施例的单向引导延伸及DSN核酸酶去偏倚的效果示意图,可见相比未做单向引导延伸及DSN处理的情况,做了单向引导延伸及DSN处理的情况能够得到较好的均一度,体现在不同位点的测序深度没有过大的差异,而未做单向引导延伸及DSN处理的情况下,不同位点的测序深度有几个数量级的差异。对应于本发明实施例的方法,本发明实施例还提供一种减少扩增偏倚的基因突变检测试剂,包括:(a)用于捕获待检测位点或待检测区域的第一步单向引导延伸引物,用于对含有目标区域DNA的接头连接产物进行预定循环数的第一步单向引导延伸;和第一通用引物,用于以第一步单向引导延伸的产物为模板进行PCR扩增,其中第一通用引物与接头连接产物上的接头序列匹配;(b)用于捕获待检测位点或区域的第二步单向引导延伸引物,用于对第一通用引物的扩增产物进行预定循环数的第二步单向引导延伸,其中第二步单向引导延伸引物相比第一步单向引导延伸引物在模板上的结合位置距离待检测位点或区域更近;和第二通用引物,用于以第二步单向引导延伸的产物为模板进行PCR扩增,其中第二通用引物与接头序列匹配;(c)双链特异性核酸酶,用于在第二通用引物的扩增产物已变性且适于退火的温度下,处理扩增产物,以减少丰度过高的DNA分子含量,增加丰度低的DNA分子的相对含量;(d)第三通用引物,用于对双链特异性核酸酶处理的产物进行PCR扩增并测序。以下通过实施例详细说明本发明的技术方案,应当理解,实施例仅是示例性的,不能理解为对本发明保护范围的限制。实施例中使用的试剂,在没有特别说明的情况下,均为市售的常规试剂。实施例本实施例对样本进行文库构建并对NRAS、KRAS、PI3KA、EGFR4个基因的7个位点进行检测,具体如下:1.含有随机单分子标签的接头设计IDX1-S:CAAGCAGAAGACGGCATACGAGATNNNNNNNNggaattaGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT(SEQIDNO:1);IDX2-S:CAAGCAGAAGACGGCATACGAGATNNNNNNNNatccggcGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT(SEQIDNO:2);IDX3-S:CAAGCAGAAGACGGCATACGAGATNNNNNNNNcaggccgGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT(SEQIDNO:3);ADT-AS:pGATCGGAAGAGC(SEQIDNO:4);ADT-AS序列5’端磷酸基团修饰。IDX1-S、IDX2-S、IDX3-S(均为接头的第一链)分别与ADT-AS序列退火成双链,构成本发明的三个接头ADT1、ADT2、ADT3。2.本实施例针对NRAS、KRAS、PIK3CA、EGFR这4个基因的常见7个突变点进行检测。针对NRAS的Q61K设计第一步单向引导延伸引物:NRAS-Q61K-STP1:TTTAATAAAAATTGAACTTCCCTCCCTCC(SEQIDNO:5);第二步单向引导延伸引物:NRAS-Q61K-STP2:TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGACTTCCCTCCCTCCCTGCCCCCTTA(SEQIDNO:6)。针对KRAS的G12D设计第一步单向引导延伸引物:KRAS-G12D-STP1:ACTGGTGGAGTATTTGATAGTGTATTAACC(SEQIDNO:7);第二步单向引导延伸引物:KRAS-G12D-STP2:TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGTTGATAGTGTATTAACCTTATGTGTGACATG(SEQIDNO:8)。针对PIK3CA的E545K设计第一步单向引导延伸引物:PIK3CA-E545K-STP1:TGACAAAGAAAGCTATATAAGATATTATTT(SEQIDNO:9);第二步单向引导延伸引物:PIK3CA-E545K-STP2:TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGTTACAGAGTAACAGACTAGCTAGAGACAATG(SEQIDNO:10)。针对EGFR的E746-A750设计第一步单向引导延伸引物:EGFR-E746-A750-STP1:CAGATCACTGGGCAGCATGTGGCAC(SEQIDNO:11);第二步单向引导延伸引物:EGFR-E746-A750-STP2:TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCATGTGGCACCATCTCACAATTGCCAGT(SEQIDNO:12)。针对EGFR的V769_D770insASV设计第一步单向引导延伸引物:EGFR-V769_D770insASV-STP1:TCAAGATCGCATTCATGCGTCTTCACCTG(SEQIDNO:13);第二步单向引导延伸引物:EGFR-V769_D770insASV-STP2:TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGGTCTTCACCTGGAAGGGGTCCATGTG(SEQIDNO:14)。针对EGFR的T790M设计第一步单向引导延伸引物:EGFR-T790M-STP1:CCTGCTGGGCATCTGCCTCACCT(SEQIDNO:15);第二步单向引导延伸引物:EGFR-T790M-STP2:TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGATCTGCCTCACCTCCACCGTGCAG(SEQIDNO:16)。针对EGFR的L858R设计第一步单向引导延伸引物:EGFR-L858R-STP1:CTGTTTCAGGGCATGAACTACTTGGAGGA(SEQIDNO:17);第二步单向引导延伸引物:EGFR-L858R-STP2:TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGGAACTACTTGGAGGACCGTCGCTTGGT(SEQIDNO:18)。所有第一步单向引导延伸引物5’端使用生物素标记修饰。所有第一步单向引导延伸引物等量混合作为第一步单向引导延伸引物组,所有第二步单向引导延伸引物等量混合作为第二步单向引导延伸引物组。预文库引物序列如下:Pre-F:GCTCTTCCGATCT(SEQIDNO:19);Uni-P1:CAAGCAGAAGACGGCATACGA(SEQIDNO:20)。最终PCR引物序列如下:Uni-P1:CAAGCAGAAGACGGCATACGA(SEQIDNO:21);Uni-P2:AATGATACGGCGACCACCGAGATCTACACTCGTCGGCAGCGTCAGATGTGTATAAGAGACAG(SEQIDNO:22)。3.对照组使用常规基因突变检测方法(直接PCR法)进行illumina测序平台文库构建:对照组实验使用2步PCR法,针对NRAS、KRAS、PIK3CA、EGFR这4个基因的常见7个突变点进行引物设计。针对NRAS的Q61K设计第一步PCR引物:NRAS-Q61K-PCR1F:ACACTCTTTCCCTACACGACGCTCTTCCGATCTACTTCCCTCCCTCCCTGCCCCCTTA(SEQIDNO:23);NRAS-Q61K-PCR1R:GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTGCCTGTCCTCATGTATTGGTCTCTCATG(SEQIDNO:24)。针对KRAS的G12D设计第一步PCR引物:KRAS-G12D-PCR1F:ACACTCTTTCCCTACACGACGCTCTTCCGATCTTTGATAGTGTATTAACCTTATGTGTGACATG(SEQIDNO:25);KRAS-G12D-PCR1R:GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTTTAGCTGTATCGTCAAGGCACTCTTGCCT(SEQIDNO:26)。针对PIK3CA的E545K设计第一步PCR引物:PIK3CA-E545K-PCR1F:ACACTCTTTCCCTACACGACGCTCTTCCGATCTTTACAGAGTAACAGACTAGCTAGAGACAATG(SEQIDNO:27);PIK3CA-E545K-PCR1R:GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTACCTGTGACTCCATAGAAAATCTTTCTCCT(SEQIDNO:28)。针对EGFR的E746-A750设计第一步PCR引物:EGFR-E746-A750-PCR1F:ACACTCTTTCCCTACACGACGCTCTTCCGATCTCATGTGGCACCATCTCACAATTGCCAGT(SEQIDNO:29);EGFR-E746-A750-PCR1R:GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTCTCACATCGAGGATTTCCTTGTTGGCT(SEQIDNO:30)。针对EGFR的V769_D770insASV设计第一步PCR引物:EGFR-V769_D770insASV-PCR1F:ACACTCTTTCCCTACACGACGCTCTTCCGATCTGTCTTCACCTGGAAGGGGTCCATGTG(SEQIDNO:31);EGFR-V769_D770insASV-PCR1R:GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTAGGTGAGGCAGATGCCCAGCAGG(SEQIDNO:32)。针对EGFR的T790M设计第一步PCR引物:EGFR-T790M-PCR1F:ACACTCTTTCCCTACACGACGCTCTTCCGATCTATCTGCCTCACCTCCACCGTGCAG(SEQIDNO:33);EGFR-T790M-PCR1R:GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTCAGGAGGCAGCCGAAGGGCATGAG(SEQIDNO:34)。针对EGFR的L858R设计第一步PCR引物:EGFR-L858R-PCR1F:ACACTCTTTCCCTACACGACGCTCTTCCGATCTGAACTACTTGGAGGACCGTCGCTTGGT(SEQIDNO:35);EGFR-L858R-PCR1R:GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTCTGCATGGTATTCTTTCTCTTCCGCAC(SEQIDNO:36)。以上对照实验组的第一步PCR引物等物质的量混在一起作为第一步PCR扩增引物组。第二步PCR引物:PCR2F:AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTAC(SEQIDNO:37);PCR2R-4:CAAGCAGAAGACGGCATACGAGAtaattctGTGACTGGAGTTCAG(SEQIDNO:38);PCR2R-5:CAAGCAGAAGACGGCATACGAGATagaggatGTGACTGGAGTTCAG(SEQIDNO:39);PCR2R-6:CAAGCAGAAGACGGCATACGAGATgagattcGTGACTGGAGTTCAG(SEQIDNO:40)。4.用于本实施例的样本为一名健康人血液基因组DNA。使用10mlEDTA抗凝管收集10ml静脉血。使用QiagenDNeasyBlood&TissueKit(250)(Qiagen69506)进行基因组DNA抽提。5.基因组DNA片段化将基因组DNA取2份(A和B)各200ng。A使用CovarisS220,超声波DNA破碎仪片段化至平均300bp大小,进行本实施例实验。样本B进行对照实验。6.片段化DNA末端修复反应体系如下表1:表1片段化DNA溶液100ngT4DNA连接酶缓冲液10μl10mMdNTP混合液4μlT4DNA聚合酶5μlT4DNA磷酸化酶5μlKlenow酶1μl总体积100μl金属浴上20℃温浴30分钟。使用120μlAmpureXPbeads进行纯化,32μl洗脱缓冲液洗脱。7.3’端加多聚腺嘌呤尾配制如下表2反应体系:表2末端修复的DNA溶液32μlKlenow酶缓冲液5μldATP10μlKlenowexo-酶3μl总体积50μl金属浴上37℃温浴30分钟。使用60μlAmpureXPbeads进行纯化,10μl洗脱缓冲液洗脱。8.连接接头配制如下表3反应体系:表33’加A的DNA溶液10μlT4DNA连接酶缓冲液25μl2μMDNA接头10μlT4DNA连接酶5μl总体积50μl金属浴上20℃温浴15分钟。使用60μlAmpureXPbeads进行纯化,34.8μl洗脱缓冲液洗脱。9.预文库扩增配制如下表4反应体系:表4PCR程序如下:a)95℃3分钟;b)3个循环程序如下:95℃15秒62℃30秒72℃30秒c)72℃5分钟d)4℃保存。使用60μlAmpureXPbeads进行纯化,50μl洗脱缓冲液洗脱。10.第一步单向引导延伸及扩增配制如下表5反应体系:表5引导延伸程序如下:a)95℃10分钟;b)20个循环程序如下:95℃30秒62℃30秒72℃1分钟c)72℃7分钟d)4℃保存。加入1ul通用引物Uni-P1(25uM)。进行第一步扩增。第一步扩增程序如下:a)95℃10分钟;b)10个循环程序如下:95℃30秒62℃30秒72℃1分钟c)72℃7分钟d)4℃保存。使用60μlAmpureXPbeads进行纯化,22μl洗脱缓冲液洗脱。11.磁珠筛选使用DynabeadsTMM-270Streptavidin(Catalognos.65305,invitrogen)磁珠对扩增产物进行筛选。12.第二步单向引导延伸及扩增配制如下表6反应体系:表6引导延伸程序如下:e)95℃10分钟;f)20个循环程序如下:95℃30秒62℃30秒72℃1分钟g)72℃7分钟h)4℃保存。加入1ul通用引物Uni-P1(25uM)。进行第二步扩增。第二步扩增程序如下:e)95℃10分钟;f)10个循环程序如下:95℃30秒62℃30秒72℃1分钟g)72℃7分钟h)4℃保存。使用60μlAmpureXPbeads进行纯化,15μl洗脱缓冲液洗脱。13.均一化处理配置如下表7预处理反应体系:表74X杂交缓冲液5ul第二步单向引导延伸及扩增产物15ul总体积20ulPCR仪上98℃2min;68℃5小时。配制如下表8的2XDSNMasterbuffer:表810XDSNMasterbuffer5ulddH2O20ul总体积25ul每个反应取22ul2XDSNMasterbuffer,PCR仪上68℃预热2小时。将22ul2XDSNMasterbuffer迅速加入到20ul预处理反应体系中,68℃10分钟。加入2ulDSN酶后68℃25分钟。加入44ul2XDSNstopsolution,轻轻吹打混匀,置冰上。使用140.8μlAmpureXPbeads进行纯化,20μl洗脱缓冲液洗脱。14.终文库扩增配置如下表9反应体系:表9均一化处理溶液20μlHIFIReadyMix(KAPABIOSYSTEMS)25μlUni-P1/Uni-P2(各5uM)5μl总体积50μlPCR程序如下:a)98℃45秒;b)10循环程序如下:98℃15秒60℃30秒72℃30秒c)72℃1分钟d)4℃保存。取其中5μl纯化产物进行2%琼脂糖凝胶电泳检测,结果如图4所示。使用60μlAmpureXPbeads进行纯化,30μl洗脱缓冲液洗脱。15.样本B进行对照实验文库构建第一步PCR扩增:配制如下表10反应体系:表10基因组DNAB200ngAmplitaqGold360MasterMix25μl第一步PCR扩增引物组(各25uM)1μlGC-增强剂1μl总体积50μl第一步扩增程序如下:a)95℃10分钟;b)5个循环程序如下:95℃30秒62℃30秒72℃1分钟c)72℃7分钟d)4℃保存。使用60μlAmpureXPbeads进行纯化,22μl洗脱缓冲液洗脱。第二步PCR扩增:配制如下表11反应体系:表11第二步扩增程序如下:a)95℃10分钟;b)5个循环程序如下:95℃30秒62℃30秒72℃1分钟c)72℃7分钟d)4℃保存。取其中5μl纯化产物进行2%琼脂糖凝胶电泳检测,结果如图4所示。使用60μlAmpureXPbeads进行纯化,22μl洗脱缓冲液洗脱。最终文库经过荧光定量PCR质控后,利用Illumina公司NextSeq500进行75bp双端测序。16.将高通量测序下机数据经过质控过滤后,进行BWA比对,并通过单分子标签进一步分析各个检测位点的测序深度,结果见下表12:表12本实施例文库A与对照文库B各位点与深度比较结果如图5所示。对比图较明显看出,本实施例的实验方法文库A中各个检测位点的测序深度较为均一。对照文库各个检测位点的测序深度严重不均一。以上内容是结合具体的实施方式对本发明所作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明。对于本发明所属
技术领域:
的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单推演或替换,都应当视为属于本发明的保护范围。SEQUENCELISTING<110>人和未来生物科技(长沙)有限公司<120>一种减少扩增偏倚的基因突变检测方法和试剂<130>16I23776<160>40<170>PatentInversion3.3<210>1<211>73<212>DNA<213>接头序列<220><221>misc_feature<222>(25)..(32)<223>nisa,c,g,ort<400>1caagcagaagacggcatacgagatnnnnnnnnggaattagtgactggagttcagacgtgt60gctcttccgatct73<210>2<211>73<212>DNA<213>接头序列<220><221>misc_feature<222>(25)..(32)<223>nisa,c,g,ort<400>2caagcagaagacggcatacgagatnnnnnnnnatccggcgtgactggagttcagacgtgt60gctcttccgatct73<210>3<211>73<212>DNA<213>接头序列<220><221>misc_feature<222>(25)..(32)<223>nisa,c,g,ort<400>3caagcagaagacggcatacgagatnnnnnnnncaggccggtgactggagttcagacgtgt60gctcttccgatct73<210>4<211>12<212>DNA<213>接头序列<400>4gatcggaagagc12<210>5<211>29<212>DNA<213>引物序列<400>5tttaataaaaattgaacttccctccctcc29<210>6<211>58<212>DNA<213>引物序列<400>6tcgtcggcagcgtcagatgtgtataagagacagacttccctccctccctgccccctta58<210>7<211>30<212>DNA<213>引物序列<400>7actggtggagtatttgatagtgtattaacc30<210>8<211>64<212>DNA<213>引物序列<400>8tcgtcggcagcgtcagatgtgtataagagacagttgatagtgtattaaccttatgtgtga60catg64<210>9<211>30<212>DNA<213>引物序列<400>9tgacaaagaaagctatataagatattattt30<210>10<211>64<212>DNA<213>引物序列<400>10tcgtcggcagcgtcagatgtgtataagagacagttacagagtaacagactagctagagac60aatg64<210>11<211>25<212>DNA<213>引物序列<400>11cagatcactgggcagcatgtggcac25<210>12<211>61<212>DNA<213>引物序列<400>12tcgtcggcagcgtcagatgtgtataagagacagcatgtggcaccatctcacaattgccag60t61<210>13<211>29<212>DNA<213>引物序列<400>13tcaagatcgcattcatgcgtcttcacctg29<210>14<211>59<212>DNA<213>引物序列<400>14tcgtcggcagcgtcagatgtgtataagagacaggtcttcacctggaaggggtccatgtg59<210>15<211>23<212>DNA<213>引物序列<400>15cctgctgggcatctgcctcacct23<210>16<211>57<212>DNA<213>引物序列<400>16tcgtcggcagcgtcagatgtgtataagagacagatctgcctcacctccaccgtgcag57<210>17<211>29<212>DNA<213>引物序列<400>17ctgtttcagggcatgaactacttggagga29<210>18<211>60<212>DNA<213>引物序列<400>18tcgtcggcagcgtcagatgtgtataagagacaggaactacttggaggaccgtcgcttggt60<210>19<211>13<212>DNA<213>引物序列<400>19gctcttccgatct13<210>20<211>21<212>DNA<213>引物序列<400>20caagcagaagacggcatacga21<210>21<211>21<212>DNA<213>引物序列<400>21caagcagaagacggcatacga21<210>22<211>62<212>DNA<213>引物序列<400>22aatgatacggcgaccaccgagatctacactcgtcggcagcgtcagatgtgtataagagac60ag62<210>23<211>58<212>DNA<213>引物序列<400>23acactctttccctacacgacgctcttccgatctacttccctccctccctgccccctta58<210>24<211>62<212>DNA<213>引物序列<400>24gtgactggagttcagacgtgtgctcttccgatctgcctgtcctcatgtattggtctctca60tg62<210>25<211>64<212>DNA<213>引物序列<400>25acactctttccctacacgacgctcttccgatctttgatagtgtattaaccttatgtgtga60catg64<210>26<211>63<212>DNA<213>引物序列<400>26gtgactggagttcagacgtgtgctcttccgatctttagctgtatcgtcaaggcactcttg60cct63<210>27<211>64<212>DNA<213>引物序列<400>27acactctttccctacacgacgctcttccgatctttacagagtaacagactagctagagac60aatg64<210>28<211>64<212>DNA<213>引物序列<400>28gtgactggagttcagacgtgtgctcttccgatctacctgtgactccatagaaaatctttc60tcct64<210>29<211>61<212>DNA<213>引物序列<400>29acactctttccctacacgacgctcttccgatctcatgtggcaccatctcacaattgccag60t61<210>30<211>61<212>DNA<213>引物序列<400>30gtgactggagttcagacgtgtgctcttccgatctctcacatcgaggatttccttgttggc60t61<210>31<211>59<212>DNA<213>引物序列<400>31acactctttccctacacgacgctcttccgatctgtcttcacctggaaggggtccatgtg59<210>32<211>57<212>DNA<213>引物序列<400>32gtgactggagttcagacgtgtgctcttccgatctaggtgaggcagatgcccagcagg57<210>33<211>57<212>DNA<213>引物序列<400>33acactctttccctacacgacgctcttccgatctatctgcctcacctccaccgtgcag57<210>34<211>58<212>DNA<213>引物序列<400>34gtgactggagttcagacgtgtgctcttccgatctcaggaggcagccgaagggcatgag58<210>35<211>60<212>DNA<213>引物序列<400>35acactctttccctacacgacgctcttccgatctgaactacttggaggaccgtcgcttggt60<210>36<211>61<212>DNA<213>引物序列<400>36gtgactggagttcagacgtgtgctcttccgatctctgcatggtattctttctcttccgca60c61<210>37<211>40<212>DNA<213>引物序列<400>37aatgatacggcgaccaccgagatctacactctttccctac40<210>38<211>45<212>DNA<213>引物序列<400>38caagcagaagacggcatacgagataattctgtgactggagttcag45<210>39<211>46<212>DNA<213>引物序列<400>39caagcagaagacggcatacgagatagaggatgtgactggagttcag46<210>40<211>46<212>DNA<213>引物序列<400>40caagcagaagacggcatacgagatgagattcgtgactggagttcag46当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 琼脂糖在pcr扩增中的应用、pcr扩增试剂盒及pcr扩增方法
- 一种基于重组酶聚合酶扩增技术原理的组织胞浆菌感染分子诊断试剂盒及其应用
- 试剂盒及其在生殖道病原体扩增检测中的用图
- 一种基于核糖体dna its1基因检测华支睾吸虫囊蚴的pcr扩增试剂盒及扩增引物的制作方法
- 一种检测简单异尖线虫和华支睾吸虫囊蚴的双重pcr扩增试剂盒及其扩增引物的制作方法
- 一种基于线粒体coi基因检测华支睾吸虫囊蚴的pcr扩增试剂盒及扩增引物的制作方法
- 一种用于血液直接荧光pcr扩增的反应液及其试剂盒的制作方法
- 同时扩增人常染色体和y染色体str基因座的荧光标记复合扩增试剂盒及其应用
- 一种人基因组dna34个基因座的复合扩增的试剂盒的制作方法
- 一种具有增强鉴别能力的常染色体str基因座荧光标记复合扩增试剂盒及其应用