一种中间体化合物以及丙硫菌唑的合成方法与流程
本发明属于有机合成领域,具体涉及一种中间体化合物以及丙硫菌唑的合成方法。
背景技术:
丙硫菌唑是一种脱甲基化抑制剂(DMIs),其作用机理是抑制真菌中甾醇的前体—羊毛甾醇l4-位脱甲基化反应。丙硫菌唑不仅具有很好的内吸活性,优异的保护、治疗和铲除活性,且持效期长。大量的田间药效试验结果表明丙硫菌唑对作物不仅具有良好的安全性,防病治病效果好,而且增产明显,同三唑类杀菌剂相比,丙硫菌唑具有更广谱的杀菌活性。
丙硫菌唑目前主要用于防治禾谷类作物如小麦、大麦、油菜、花生、水稻和豆类作物等众多病害。丙硫菌唑几乎对所有麦类病害都有很好的防治效果,如小麦和大麦的白粉病、纹枯病、枯萎病、叶斑病、锈病、菌核病、网斑病、云纹病等。丙硫菌唑还能防治油菜和花生的土传病害,如菌核病,以及主要叶面病害,如灰霉病、黑斑病、褐斑病、黑胫病和锈病等。
根据硫原子的来源不同,丙硫菌唑的制备策略可分为两大类。第一类制备丙硫菌唑的策略是以羟基三唑化合物为关键中间体,与硫磺反应得丙硫菌唑(US4913727)。硫磺在这类反应中作为丙硫菌唑化合物硫原子的来源。该方法的关键中间体可以氯化物(US4913727)或环氧化合物(US5146001)为起始原料和三氮唑经取代反应制得。此取代反应会同时产生相当量的区域异构体,需要通过精制去除,导致收率欠佳(51~53%)。关键中间体也可以氯代酮为原料和三氮唑反应,再和格氏试剂反应制得,此方法同样存在区域选择性问题。
美国专利US5789430公开了以化合物和硫磺直接反应制备丙硫菌唑的方法。该反应以N-甲基吡咯酮为溶剂在200℃下反应44小时得丙硫菌唑,收率为20%。US5789430同时也公开了以化合物和硫磺反应制备丙硫菌唑的改进方法。该改进方法是将化合物在THF溶剂里先用n-BuLi拔氢,再和硫磺反应,所得丙硫菌唑的收率大大提高(93%),但该技术方案需要无水无氧和超低温反应设备和条件,同时需要使用大于两当量的高危险性的n-BuLi试剂,使该技术方案成本高,且操作不安全,不利于工业化生产。
另外,该技术方案同时也受自身化学区域选择性的困扰,例如:(1)在关键中间体用n-BuLi拔氢过程中若控制不当将导致区域异构杂质11的产生;(2)在制备关键中间体时如果区域异构体不被完全分离提纯干净,将导致区域异构杂质的产生。这些高要求的分离提纯不仅产生大量三废,同时大大增加成本。
US2013005985公开了一种使用格氏试剂如i-PrMgCl代替n-BuLi对化合物进行拔氢后再硫化制备丙硫菌唑的方法。该方法解决了使用n-BuLi试剂危险性问题,但该技术方案还是需要无水无氧和超低温反应设备和条件,同时需要使用大于两当量的格氏试剂,并且产率大幅下降(从n-BuLi的93%下降到68%)。
技术实现要素:
本部分的目的在于概述本发明的实施例的一些方面以及简要介绍一些较佳实施例。在本部分以及本申请的说明书摘要和发明名称中可能会做些简化或省略以避免使本部分、说明书摘要和发明名称的目的模糊,而这种简化或省略不能用于限制本发明的范围。
鉴于上述和/或现有合成丙硫菌唑的技术空白,提出了本发明。
因此,本发明其中的一个目的是提供一种中间体化合物,该中间体化合物能够用于丙硫菌唑的合成。
为解决上述技术问题,本发明提供了如下技术方案:一种中间体化合物,其化学结构式为:
本发明其中的另一个目的是解决现有技术中丙硫菌唑合成方法的不足,提高每一步的转化率,减少副产物,简化反应条件,降低能耗,缩短反应时间,提高原子利用率,减少三废。
为解决上述技术问题,本发明提供了如下技术方案:一种丙硫菌唑的合成方法,包括,以1,2,4-三氮唑为原料经硫氧化得到巯基-1,2,4-三氮唑;巯基-1,2,4-三氮唑经氧化形成5,5’-二硫基-双(1,2,4-三氮唑);与2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇发生取代反应得到如权利要求1所述中间体化合物;经还原得到目标产物丙硫菌唑。
作为本发明所述丙硫菌唑的合成方法的一种优选方案,其中:所述以1,2,4-三氮唑为原料经硫氧化得到巯基-1,2,4-三氮唑,包括,将1,2,4-三氮唑溶于有机溶剂中,加入同1,2,4-三氮唑摩尔比为1:3~1:6的硫,加热至100~180℃,反应2~8h,,冷却至室温,过滤,滤液用饱和氯化钠洗涤,经乙酸乙酯萃取,分出有机相,干燥,蒸除乙酸乙酯得巯基-1,2,4-三氮唑;其中,有机溶剂为DMF或甲苯或DMSO中一种或多种;其中,干燥采用固体干燥剂为无水硫酸镁、无水氯化钙、无水硫酸钠、无水硫酸钙或活性氧化铝中的一种或几种。
作为本发明所述丙硫菌唑的合成方法的一种优选方案,其中:所述加热至100~180℃,其温度优选为110~160℃。
作为本发明所述丙硫菌唑的合成方法的一种优选方案,其中:所述反应2~8h,其反应时间优选为3~6h。
作为本发明所述丙硫菌唑的合成方法的一种优选方案,其中:所述巯基-1,2,4-三氮唑经氧化形成5,5’-二硫基-双(1,2,4-三氮唑),包括,将巯基-1,2,4-三氮唑溶于DCM中,加入吡啶,控制温度为-2℃~6℃,搅拌,加入苯磺酰氯,反应后去除DCM,剩余物加入水和乙酸乙酯,反应后过滤,并用水和乙酸乙酯洗涤,将过滤得到的固体产物干燥,得到固体化合物(Ⅴ)即5,5’-二硫基-双(1,2,4-三氮唑);其中,干燥采用固体干燥剂为无水硫酸镁、无水氯化钙、无水硫酸钠、无水硫酸钙或活性氧化铝中的一种或几种。
作为本发明所述丙硫菌唑的合成方法的一种优选方案,其中:所述反应温度优选为0℃~4℃。
作为本发明所述丙硫菌唑的合成方法的一种优选方案,其中:所述与2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇发生取代反应,包括,将所述固体化合物(Ⅴ)和有机溶剂、碳酸钾搅拌混合,加热至20℃~100℃,加入2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇中间体化合物,反应后冷却至室温,抽滤,滤液用饱和食盐水洗涤,乙酸乙酯萃取,有机相用饱和食盐水洗涤,分出有机相,干燥,旋蒸得到化合物(Ⅵ);其中,有机溶剂为甲醇、乙醇、异丙醇、乙腈、丙酮或DMF中的一种或几种;其中,固体化合物(V)同碳酸钾摩尔比为1:2~1:4;其中,固体化合物(V)同2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇化合物(Ⅱ)摩尔比为1:2~1:5;其中,干燥采用固体干燥剂为无水硫酸镁、无水氯化钙、无水硫酸钠、无水硫酸钙或活性氧化铝中的一种或几种。
作为本发明所述丙硫菌唑的合成方法的一种优选方案,其中:所述固体干燥剂,优选为无水硫酸钠。
作为本发明所述丙硫菌唑的合成方法的一种优选方案,其中:所述加热至20℃~100℃,优选40℃~80℃。
作为本发明所述丙硫菌唑的合成方法的一种优选方案,其中:所述有机溶剂为,优选为DMF。
作为本发明所述丙硫菌唑的合成方法的一种优选方案,其中:所述经还原得到目标产物丙硫菌唑,包括,将化合物(Ⅵ)溶于有机溶剂中,加入还原剂,反应温度20℃-60℃条件下搅拌反应后,重结晶,析出晶体过滤得目标化合物丙硫菌唑。其中,有机溶剂为甲醇、乙醇、异丙醇、乙腈、丙酮中一种或多种;其中,还原剂为TCEP、DTT、Zn、硼氢化钠、氢化铝锂中的一种或几种。
作为本发明所述丙硫菌唑的合成方法的一种优选方案,其中:所述有机溶剂,优选为甲醇。
作为本发明所述丙硫菌唑的合成方法的一种优选方案,其中:所述反应温度,优选为30℃~50℃。
作为本发明所述丙硫菌唑的合成方法的一种优选方案,其中:所述还原剂,优选为金属Zn。
本发明的有益效果:
(1)本发明合成工艺转化率和选择性高,合成原料便宜易得,降低了生产成本。
(2)反应条件温和易控,操作简便,产品提纯容易,可以直接重结晶得到产物。
(3)各步中间体控制方法简单、准确,产品收率较高,原子经济性较好,避免繁琐的后处理,具有很大的竞争优势和工业生产利用价值。
(4)避免了使用强碱等原料,三废极低,符合绿色化学的理念。
附图说明
为了更清楚地说明本发明实施例的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其它的附图。其中:
图1为化合物Ⅳ的MS图谱;
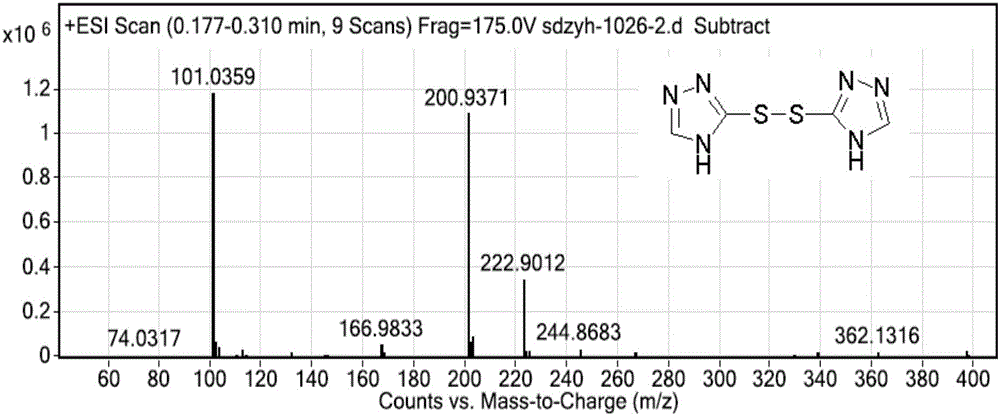
图2为化合物Ⅴ的MS图谱;
图3为化合物Ⅵ的MS图谱;
图4为化合物Ⅶ的MS图谱;
图5为化合物Ⅶ的1H-NMR图谱,其中,1H NMR(400MHz,Chloroform-d)δ11.92(s,1H),7.85(s,1H),7.59–7.50(m,1H),7.40–7.33(m,1H),7.21(tt,J=7.3,5.3Hz,2H),4.79(d,J=14.6Hz,1H),4.50(d,J=14.6Hz,1H),4.27(s,1H),3.62(d,J=14.0Hz,1H),3.17(d,J=14.0Hz,1H),1.02–0.85(m,2H),0.85–0.73(m,2H)。
具体实施方式
为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合具体实施例对本发明的具体实施方式做详细的说明。
在下面的描述中阐述了很多具体细节以便于充分理解本发明,但是本发明还可以采用其他不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似推广,因此本发明不受下面公开的具体实施例的限制。
其次,此处所称的“一个实施例”或“实施例”是指可包含于本发明至少一个实现方式中的特定特征、结构或特性。在本说明书中不同地方出现的“在一个实施例中”并非均指同一个实施例,也不是单独的或选择性的与其他实施例互相排斥的实施例。
为了描述本发明的方便,使用各种试剂的常规和非常规缩写。这些缩写是本领域技术人员熟悉的,但是为了清楚在下面列出:
THF:四氢呋喃
DMF:N,N-二甲基甲酰胺
DMSO:二甲基亚砜
DCM:二氯甲烷
TCEP:三(2-羧乙基)膦
DTT:二硫苏糖醇
实施例1:
2-(1-氯环丙基)-3-氯-1-(2-氯苯基)-2-丙醇的合成
在有恒压滴定漏斗和冷凝管的250mL圆底三口烧瓶装置下,进行无水无氧操作氮气保护条件下,将10g(0.062mol)2-氯氯苄与50mLTHF溶液混合置于恒压滴液漏斗中,在三口烧瓶中加入1.8g(0.074mol)镁屑和10ml的THF溶液和少量的碘,滴入2ml 2-氯氯苄的THF溶液,微热引发,将装置放入冰浴中缓慢滴加(滴/3s)直至滴完,滴完后再反应1h,将上述格氏反应物慢慢滴入9.0g(0.058mol)1-氯-1-氯乙酰基环丙烷的THF(30mL)溶液缓慢滴加(滴/3s)直至滴完,滴完继续反应1h,装置一直放在冰浴中。最后缓慢将反应液缓慢加入到饱和的NH4Cl冰水溶液中淬灭反应,搅拌1h,用分液漏斗分出有机相,加入无水硫酸钠干燥后,蒸除THF得到,得14.45g化合物(Ⅱ)浅黄色油状液体,收率为89%。
实施例2:
丙硫菌唑的合成
化合物(Ⅳ)的合成
将5g(0.072mol)1,2,4-三氮唑溶于15ml的DMF中,加入6.9g(0.216mol)升华硫,加热到140℃,反应3.5h后,冷却至室温,过滤,滤液用饱和氯化钠洗涤,乙酸乙酯萃取,分出有机相,用无水硫酸钠干燥,蒸除乙酸乙酯得固体化合物(Ⅳ)6.95g,收率为95%。
化合物(Ⅴ)的合成
将3.03g(30mmol)化合物(Ⅳ)溶于30mLDCM中,加入2.37g(30mmol)重蒸吡啶。冰浴搅拌条件下,滴加2.64g(15mmol)苯磺酰氯,1h左右滴加完毕。移去冰水浴,室温下搅拌5h。蒸去DCM,剩余物加入15mL水和10mL乙酸乙酯,反应1h,过滤,并用适量的水和乙酸乙酯洗涤。将过滤得到的固体产物在真空干燥箱里抽干,得到固体化合物(Ⅴ)2.88g,收率为96%。
化合物(Ⅵ)的合成
将4g(20mmol)化合物(Ⅴ)和20ml DMF,5.52g(40mmol)碳酸钾搅拌混合,加热至50℃。滴加化合物(Ⅱ)11.74g(42mmol),2h滴加完毕,继续反应h,停止反应,冷却至室温。抽滤,滤液用饱和食盐水洗涤,乙酸乙酯萃取,有机相用饱和食盐水洗涤三次,分出有机相,无水硫酸钠干燥,旋蒸的产物。得到化合物(Ⅵ)11.4g,收率为83%。
目标化合物(Ⅶ)丙硫菌唑的合成
取5g(0.0072mol)化合物(Ⅵ)溶于30ml的甲醇中,加入0.97g(0.015mol)金属Zn,30℃搅拌反应3h,停止反应,直接重结晶,析出晶体过滤得目标化合物(Ⅶ)丙硫菌唑4.3g,收率为86%。
本发明在上述实施例方式中,重新设计合成路线来降低生产成本,提高每一步的转化率,其以1,2,4-三氮唑为原料经硫氧化得到巯基-1,2,4-三氮唑,再经过氧化形成二硫键,再与由1-氯-1-氯乙酰基环丙烷与2-氯氯苄经格式反应制得的2-(1-氯环丙烷)-3-氯-1-(2-氯苯基)-2-丙醇发生取代反应,再经还原就得到目标产物丙硫菌唑。其原理通过化学式表示为:
对比实施例1:
丙硫菌唑的合成
化合物(Ⅳ)的合成
将5g(0.072mol)1,2,4-三氮唑溶于15ml的DMSO中,加入11.52g(0.36mol)升华硫,加热到180℃,反应5h后,冷却至室温,过滤,滤液用饱和氯化钠洗涤,乙酸乙酯萃取,分出有机相,用无水硫酸钠干燥,蒸除乙酸乙酯得固体化合物(Ⅳ)5.89g,收率为81%。
化合物(Ⅴ)的合成
将3.03g(30mmol)化合物(Ⅳ)溶于30mLDCM中,加入3.16g(40mmol)重蒸吡啶。6℃搅拌条件下,滴加3.53g(20mmol)苯磺酰氯,1h左右滴加完毕。室温下搅拌4h。蒸去DCM,剩余物加入20mL水和20mL乙酸乙酯,反应1h,过滤,并用适量的水和乙酸乙酯洗涤。将过滤得到的固体产物在真空干燥箱里抽干,得到固体化合物(Ⅴ)2.49g,收率为83%。
化合物(Ⅵ)的合成
将4g(20mmol)化合物(Ⅴ)和40ml乙腈,8.,28g(60mmol)碳酸钾搅拌混合,加热至80℃。滴加化合物(Ⅱ)21.24g(80mmol),2h滴加完毕,继续反应4h,停止反应,冷却至室温。抽滤,滤液用饱和食盐水洗涤,乙酸乙酯萃取,有机相用饱和食盐水洗涤三次,分出有机相,无水硫酸钠干燥,旋蒸的产物。得到化合物(Ⅵ)10.7g,收率为78%。
目标化合物(Ⅶ)丙硫菌唑的合成
取5g(0.0072mol)化合物(Ⅵ)溶于30ml的乙醇中,加入1.98g(0.0079mol)TCEP,50℃搅拌反应3h,停止反应,直接重结晶,析出晶体过滤得目标化合物(Ⅶ)丙硫菌唑4.1g,收率为82%。
对比实施例2:
丙硫菌唑的合成
化合物(Ⅳ)的合成
将5g(0.072mol)1,2,4-三氮唑溶于20ml的甲苯中,加入9.22g(0.288mol)升华硫,加热到100℃,反应10h后,冷却至室温,过滤,滤液用饱和氯化钠洗涤,乙酸乙酯萃取,分出有机相,用无水硫酸钠干燥,蒸除乙酸乙酯得固体化合物(Ⅳ)4.07g,收率为56%。
化合物(Ⅴ)的合成
将3.03g(30mmol)化合物(Ⅳ)溶于30mLDCM中,加入3.16g(40mmol)重蒸吡啶。-2℃搅拌条件下,滴加3.53g(20mmol)苯磺酰氯,1h左右滴加完毕。室温下搅拌5h。蒸去DCM,剩余物加入20mL水和20mL乙酸乙酯,反应1h,过滤,并用适量的水和乙酸乙酯洗涤。将过滤得到的固体产物在真空干燥箱里抽干,得到固体化合物(Ⅴ)2.1g,收率为70%。
化合物(Ⅵ)的合成
将4g(20mmol)化合物(Ⅴ)和40ml丙酮,11.04g(80mmol)碳酸钾搅拌混合,加热至30℃。滴加化合物(Ⅱ)21.24g(60mmol),2h滴加完毕,继续反应4h,停止反应,冷却至室温。抽滤,滤液用饱和食盐水洗涤,乙酸乙酯萃取,有机相用饱和食盐水洗涤三次,分出有机相,无水硫酸钠干燥,旋蒸的产物。得到化合物(Ⅵ)7.0g,收率为51%。
目标化合物(Ⅶ)丙硫菌唑的合成
取5g(0.0072mol)化合物(Ⅵ)溶于30ml的丙酮中,加入0.55g(0.0144mol)硼氢化钠,20℃搅拌反应5h,停止反应,直接重结晶,析出晶体过滤得目标化合物(Ⅶ)丙硫菌唑2.72g,收率为55%。
实施例3
化合物(Ⅵ)的合成
将6g(30mmol)化合物(Ⅴ)和30ml DMF,12.42g(90mmol)碳酸钾搅拌混合,加热至60℃。滴加化合物(Ⅱ)17.61g(63mmol),2h滴加完毕,继续反应,停止反应,冷却至室温。抽滤,滤液用饱和食盐水洗涤,乙酸乙酯萃取,有机相用饱和食盐水洗涤三次,分出有机相,无水硫酸钠干燥,旋蒸的产物。得到化合物(Ⅵ)17.31g,收率为84%。
目标化合物(Ⅶ)丙硫菌唑的合成
取6.25g(0.009mol)化合物(Ⅵ)溶于30ml的甲醇中,加入1.21g(0.019mol)金属Zn,40℃搅拌反应3h,停止反应,直接重结晶,析出晶体过滤得目标化合物(Ⅶ)丙硫菌唑5.5g,收率为88%。
实施例4
化合物(Ⅵ)的合成
将6g(30mmol)化合物(Ⅴ)和30ml DMF,12.42g(90mmol)碳酸钾搅拌混合,加热至80℃。滴加化合物(Ⅱ)17.61g(63mmol),2h滴加完毕,继续反应,停止反应,冷却至室温。抽滤,滤液用饱和食盐水洗涤,乙酸乙酯萃取,有机相用饱和食盐水洗涤三次,分出有机相,无水硫酸钠干燥,旋蒸的产物。得到化合物(Ⅵ)16.49g,收率为80%。
目标化合物(Ⅶ)丙硫菌唑的合成
取6.25g(0.009mol)化合物(Ⅵ)溶于30ml的甲醇中,加入0.97g(0.015mol)金属硼氢化钠,50℃搅拌反应3h,停止反应,直接重结晶,析出晶体过滤得目标化合物(Ⅶ)丙硫菌唑5.06g,收率为81%。
实施例5
化合物(Ⅵ)的合成
将6g(30mmol)化合物(Ⅴ)和30ml DMF,12.42g(90mmol)碳酸钾搅拌混合,加热至100℃。滴加化合物(Ⅱ)17.61g(63mmol),2h滴加完毕,继续反应,停止反应,冷却至室温。抽滤,滤液用饱和食盐水洗涤,乙酸乙酯萃取,有机相用饱和食盐水洗涤三次,分出有机相,无水硫酸钠干燥,旋蒸的产物。得到化合物(Ⅵ)11.54g,收率为56%。
目标化合物(Ⅶ)丙硫菌唑的合成
取6.25g(0.009mol)化合物(Ⅵ)溶于30ml的甲醇中,加入0.97g(0.015mol)金属TECP,20℃搅拌反应3h,停止反应,直接重结晶,析出晶体过滤得目标化合物(Ⅶ)丙硫菌唑3.44g,收率为55%。
实施例3~5反应原理为:
在上述实施例1~5中,产生了合成目标化合物的关键中间体化合物,其化学结构式为:
基于上述实施例所提供的数据,可知本发明提供的丙硫菌唑的合成方法,能够得到较为优异的收率。
在具体的实施例中,实施例1和2,均采用本发明提供的关于有机溶剂选择、温度控制范围及还原剂选择的优选方案,故而在每一阶段目标化合物的收率均较为优异。相比之下,对比实施例1,基本遵循优选方案,而在合成路线中,个别目标化合物的合成中未采用本发明提供的温度或还原剂的优选方案,相应得率均不理想。对比实施例2中,每一步目标化和物的合成,均未采用本发明关于反应时间、反应温度、有机溶剂和还原剂选择的优选范围。
其中具体机理如下:
从反应温度和反应时间角度看,过高的反应温度及过长的反应时间,会出现反应平衡目标产物占主要比例时,反应仍在继续,平衡进一步推进,使得副产物增多;过低的反应温度及过短的反应时间,会使得反应无法充分进行,并不能呈现较好的化学平衡,继而获得较佳的目标产物比收率。本发明通过对合成路线的设计,及每一步目标产物合成的反应温度和反应时间的匹配设计,给出优选的控制范围。
应说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的精神和范围,其均应涵盖在本发明的权利要求范围当中。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种丙辛吲哚盐酸盐药物中间体丙辛吲哚的合成方法与制造工艺
- 一种吲哚洛尔药物中间体(一)‑4‑(2‑羟基‑3‑异丙氨基丙氧)吲哚的合成方法与制造工艺
- 依替米贝的中间体及其合成方法与依替米贝的合成方法与制造工艺
- 一种白消安药物中间体1,4‑双甲基磺酸丁二酯的合成方法与制造工艺
- 一种塞利洛尔药物中间体3‑(4‑乙氧基苯基)‑1,1‑二乙基脲的合成方法与制造工艺
- 一种羟基保泰松药物中间体4‑羟基偶氮苯的合成方法与制造工艺
- 一种甲氧卡那霉素药物中间体γ‑氨基‑α‑羟基丁酸的合成方法与制造工艺
- 一种非那西丁药物中间体对硝基苯乙醚的合成方法与制造工艺
- 一种间尼索地平药物中间体乙酰乙酸异丁酯的合成方法与制造工艺
- 一种甲氧苄啶药物中间体3,4,5—三甲氧基苯甲酸甲酯的合成方法与制造工艺
- 依替米贝的中间体及其合成方法与依替米贝的合成方法与制造工艺
- 一种白消安药物中间体1,4‑双甲基磺酸丁二酯的合成方法与制造工艺
- 一种塞利洛尔药物中间体3‑(4‑乙氧基苯基)‑1,1‑二乙基脲的合成方法与制造工艺
- 一种羟基保泰松药物中间体4‑羟基偶氮苯的合成方法与制造工艺
- 一种甲氧卡那霉素药物中间体γ‑氨基‑α‑羟基丁酸的合成方法与制造工艺
- 一种非那西丁药物中间体对硝基苯乙醚的合成方法与制造工艺
- 一种间尼索地平药物中间体乙酰乙酸异丁酯的合成方法与制造工艺
- 一种甲氧苄啶药物中间体3,4,5—三甲氧基苯甲酸甲酯的合成方法与制造工艺
- 一种氯苯达诺药物中间体邻氯苯甲酸的合成方法与制造工艺
- 一种噢哢类抗癌化合物药物中间体2,4,6‑三羟基苯乙酮的合成方法与制造工艺