氮杂二环式化合物的结晶的制作方法
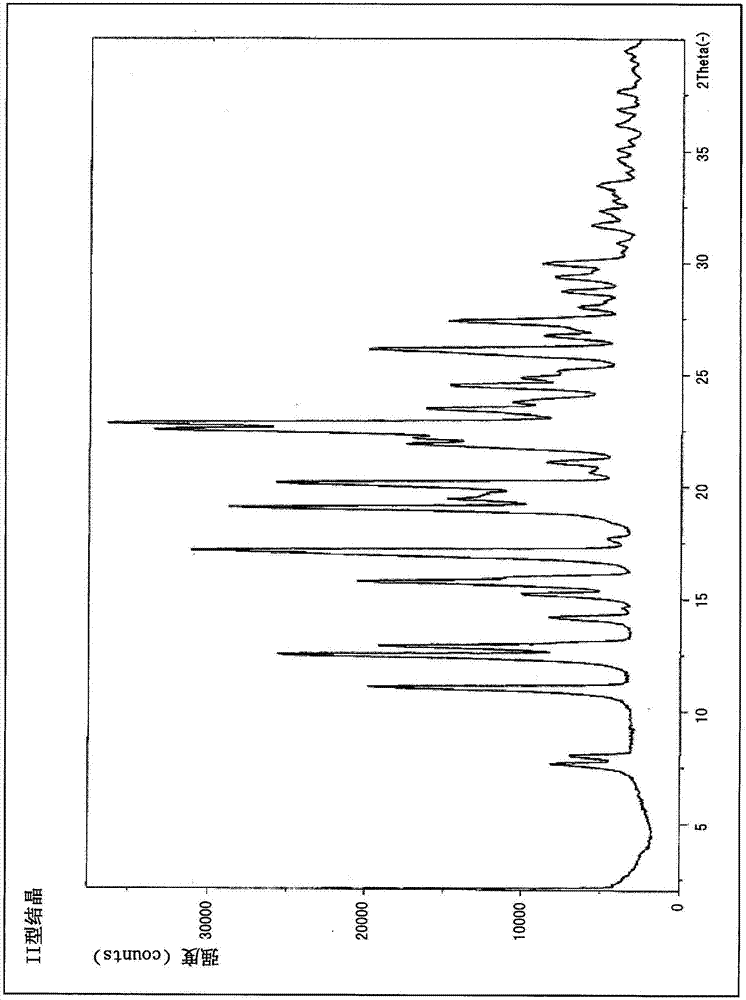
本发明涉及稳定且口服吸收性优异、作为抗肿瘤剂有用的氮杂二环式化合物的新型的结晶。
背景技术:
:通常,作为医药品的有效活性成分使用化合物时,为了稳定地保持品质和/或使保管管理变得容易,需要化合物的化学上的稳定性和物理学上的稳定性。因此,优选得到的化合物为稳定的晶型,通常,选择最稳定晶型作为医药品用的原料药的先例很多。目前,作为抗肿瘤剂,报道了多种hsp90抑制剂,在专利文献1和2中,作为具有优异的hsp90抑制作用且表现出抗肿瘤活性的化合物,记载了3-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺(以下也称为“化合物1”)。另一方面,通常情况下,在口服给药用的医药组合物中,除了有效成分的稳定性之外,还需求口服给药时的优异的吸收性,专利文献1和2完全没有关于化合物1结晶、以及该结晶的稳定性和口服吸收性的记载。现有技术文献专利文献专利文献1:国际公开第2012/093708号专利文献2:国际公开第2011/004610号技术实现要素:发明要解决的技术问题本发明的目的在于提供一种作为抗肿瘤剂有用的化合物1的稳定且口服吸收性优异的结晶。用于解决技术问题的手段本发明的发明人为了解决上述技术问题,在按照专利文献1中记载的制造方法合成化合物1时,得到了化合物1的i型结晶。但是,如后述的实施例所述,i型结晶的口服吸收性存在问题,因而进一步反复对结晶化条件进行了研究。结果发现通过将化合物1添加到特定的有机溶剂中使其悬浊,能够得到ii型结晶,并且发现ii型结晶与i型结晶相比,稳定性和口服吸收性优异,从而完成了本发明。即,本发明提供下述〔1〕~〔15〕。〔1〕一种3-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的ii型结晶,其特征在于:在粉末x射线衍射图谱中具有选自衍射角(2θ±0.2°)7.7°、8.0°、11.1°、12.5°、12.9°、15.2°、15.8°、17.2°、19.0°、22.5°、26.1°和27.4°中的至少3个以上的特征峰。〔2〕如〔1〕所记载的ii型结晶,其为在粉末x射线衍射图谱中具有选自衍射角(2θ±0.2°)7.7°、8.0°、11.1°、12.5°、12.9°、15.2°、15.8°、17.2°、19.0°、22.5°、26.1°和27.4°中的至少5个以上的特征峰的结晶。〔3〕如〔1〕或〔2〕所记载的ii型结晶,通过差热-热重同时测定确定的吸热峰在270℃附近。〔4〕一种含有〔1〕~〔3〕中任一项所记载的ii型结晶的医药组合物。〔5〕一种含有〔1〕~〔3〕中任一项所记载的ii型结晶的口服给药用医药组合物。〔6〕一种含有〔1〕~〔3〕中任一项所记载的ii型结晶的抗肿瘤剂。〔7〕〔1〕~〔3〕中任一项所记载的ii型结晶在制造医药组合物中的使用。〔8〕如〔7〕所记载的使用,其中,医药组合物是口服给药用医药组合物。〔9〕〔1〕~〔3〕中任一项所记载的ii型结晶在制造抗肿瘤剂中的使用。〔10〕如〔1〕~〔3〕中任一项所记载的ii型结晶,其作为医药使用。〔11〕如〔1〕~〔3〕中任一项所记载的ii型结晶,其用于肿瘤的治疗。〔12〕一种肿瘤的治疗方法,其包括向需要进行肿瘤治疗的对象给予有效量的〔1〕~〔3〕中任一项所记载的ii型结晶的步骤。〔13〕一种〔1〕~〔3〕中任一项所记载的ii型结晶的制造方法,包括:(1)将3-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺在有机溶剂中加热悬浊,得到悬浊液的工序;和(2)由上述(1)中得到的悬浊液获得固态的3-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的工序。〔14〕如〔13〕所记载的ii型结晶的制造方法,其中,有机溶剂为2-丙醇、乙酸甲酯、乙酸乙酯、乙酸丙酯、乙酸丁酯、环戊基甲基醚、甲乙酮、甲基异丁基酮、丙酮、乙腈或它们的混合溶剂。〔15〕一种〔1〕~〔3〕中任一项所记载的ii型结晶的制造方法,包括:(1)将3-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺在选自2-丙醇、乙酸甲酯、乙酸乙酯、乙酸丙酯、乙酸丁酯、环戊基甲基醚、甲乙酮、甲基异丁基酮、丙酮、乙腈以及它们的混合溶剂中的有机溶剂中悬浊,得到悬浊液的工序;和(2)由上述(1)中得到的悬浊液获得固态的3-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的工序。发明效果根据本发明,化合物1的ii型结晶具有高稳定性和优异的口服吸收性,作为口服用医药品有用。附图说明图1表示化合物1的i型结晶的粉末x射线衍射图谱(纵轴表示强度(cps)、横轴表示衍射角(2θ±0.2°))。图2表示化合物1的ii型结晶的粉末x射线衍射图谱(纵轴表示强度(cps)、横轴表示衍射角(2θ±0.2°))。图3表示化合物1的ii型结晶的差示扫描量热(dsc)曲线。图4表示化合物1的ii型结晶的血中浓度测定试验结果。具体实施方式本发明的化合物1是3-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺(下述式)。已知化合物1具有hsp90抑制活性,表现出优异的抗肿瘤活性。其中,化合物1可以基于专利文献1和2中记载的制造方法合成。本发明的结晶只要含有化合物1的ii型结晶即可,既可以是ii型结晶的单一结晶,也可以是含有ii型结晶以外的结晶的多晶型混合物。在本发明的结晶中,优选高纯度的ii型结晶。具体而言,优选结晶的化学纯度在90%以上的ii型结晶,更优选在95%以上,特别优选在98%以上。本发明的ii型结晶能够通过将化合物1添加到特定的有机溶剂中使其悬浊而获得。具体而言,能够通过包括下述工序(1)和(2)的制造方法获得ii型结晶:(1)将化合物1在有机溶剂中悬浊,得到悬浊液的工序,(2)由上述(1)中得到的悬浊液获得固态的化合物1的工序。在此,作为在有机溶剂中添加的化合物1,不管是否为结晶均可,但从得到高纯度的ii型结晶的观点考虑,优选使用结晶的化合物1,特别优选使用化合物1的ii型结晶。并且,在本发明的结晶化时可以使用晶种。从得到高纯度的ii型结晶的观点考虑,优选ii型结晶作为晶种。作为本发明的结晶化中使用的有机溶剂,可以例示:甲醇、正丙醇、2-丙醇、乙二醇等醇类;乙酸甲酯、乙酸乙酯、乙酸丙酯、乙酸丁酯等脂肪族羧酸酯类;二乙基醚、甲基叔丁基醚、环戊基甲基醚、1,4-二噁烷、四氢呋喃等醚类;丙酮、甲乙酮、甲基异丁基酮、环己酮等酮类;甲苯、二甲苯、氯苯等芳香族溶剂;乙腈、n-甲基-2-吡咯烷酮、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、1,3-二甲基-2-咪唑啉酮和二甲基亚砜等非质子型极性有机溶剂或它们的混合溶剂。优选为酮类、碳原子数3以上的一元醇、二元醇、脂肪族羧酸酯类、醚类、非质子型极性有机溶剂或它们的混合溶剂,进一步优选为2-丙醇、乙酸甲酯、乙酸乙酯、乙酸丙酯、乙酸丁酯、环戊基甲基醚、甲乙酮、甲基异丁基酮、丙酮、乙腈或它们的混合溶剂。从ii型结晶的纯度和收率的观点考虑,特别优选为乙酸甲酯、甲乙酮、丙酮或它们的混合溶剂。关于本发明的结晶化时有机溶剂的量(v/w),从ii型结晶的纯度和收率的观点考虑,相对于化合物1的量优选为2~30倍量,更优选为3~20倍量,特别优选为4~15倍量。在本发明的结晶化时,优选将化合物1添加到有机溶剂中加热悬浊,并进行长时间回流。本发明的结晶化时的加热温度只要是能够回流的温度即可,没有特别限定,可以根据所使用的有机溶剂适当设定。优选为52℃~126℃。关于本发明的结晶化时进行回流的时间,在过短时结晶化不能充分进行,无法获得高纯度的结晶;而在过长时发生结晶的分解,收率下降,因而优选为12~60小时,更优选为16~48小时。在本发明的结晶化时,通过在回流后进行冷却能够获得析出的ii型结晶。作为冷却温度可以适时设定,但优选为室温。所析出的结晶例如可以通过过滤、利用有机溶剂的清洗、减压干燥等公知的分离精制手段从上述溶解溶液或混合溶液分离精制。作为清洗所使用的有机溶剂,例如可以列举低级醇、丙酮、乙腈等。也可以使用在ii型结晶化中使用的有机溶剂进行清洗。如上所述操作得到的本发明的ii型结晶为如图2所示在粉末x射线衍射图谱中具有选自衍射角(2θ±0.2°)7.7°、8.0°、11.1°、12.5°、12.9°、15.2°、15.8°、17.2°、19.0°、22.5°、26.1°和27.4°中的3个以上、优选5个以上、更优选8个以上、进一步优选12个特征峰的结晶。另外,如图3所示的差示扫描量热测定(dsc测定)的结果所示,ii型结晶在270℃附近具有吸热峰。相对于此,i型结晶是如图1所示在粉末x射线衍射图谱中在衍射角(2θ±0.2°)8.1°、12.1°、14.0°、16.2°、21.5°、25.4°和28.3°具有特征峰的结晶。粉末x射线衍射图谱中的峰值有时因测定仪器或峰的读取条件等测定条件会出现少许误差。本说明书中的峰值可以在±0.2°左右的范围内具有测定误差。在dsc测定中,所测得的吸热峰(峰顶值)的测定温度有时会因每1分钟的升温幅度或试样量以及纯度等而发生变化。本说明书中的“附近”的术语表示±5.0℃。如后述的实施例,化合物1的i型结晶与ii型结晶的口服给药时的吸收性有很大差异。通常情况下,亚稳型的溶解度比稳定型的溶解度高(辻彰著、新药剂学、南江堂),因而稳定型的ii型结晶的口服吸收性比亚稳型的i型结晶高是预料不到的结果。另外,化合物1的i型结晶通过进行回流转变为ii型结晶。于是,ii型结晶在高温条件和高湿条件下稳定。因此,ii型结晶比i型结晶稳定,作为医药品原料有用。因此,本发明的ii型结晶作为医药组合物的有效成分有用,特别是作为口服给药用医药组合物的有效成分有用。由于化合物1具有优异的hsp90抑制活性,所以本发明的ii型结晶作为抗肿瘤剂有用。作为对象的癌没有特别限制,可以列举头颈癌、消化器官癌(食道癌、胃癌、消化道间质瘤、十二指肠癌、肝癌、胆道癌(胆囊·胆管癌等)、胰腺癌、小肠癌、大肠癌(结肠直肠癌、结肠癌、直肠癌等)等)、肺癌、乳腺癌、卵巢癌、子宫癌(子宫颈癌、子宫体癌等)、肾癌、膀胱癌、前列腺癌、尿路上皮癌、骨·软组织肉瘤、血液癌(b细胞淋巴肿瘤、慢性淋巴性白血病、末梢性t细胞性淋巴肿瘤、骨髓增生异常综合征、急性骨髄性白血病、急性淋巴性白血病等)、多发性骨髄肿瘤、皮肤癌、间皮瘤等。在将本发明的ii型结晶作为医药组合物的有效成分使用时,可以根据需要配合药学上可接受的载体,可以根据预防或治疗目的采用各种给药剂型,作为该剂型,优选片剂、胶囊剂、颗粒剂、细粒剂、散剂等口服剂。它们的给药形态可以通过各所属
技术领域:
的技术人员公知常用的制剂方法制造。实施例以下,列举实施例对本发明进行更具体的说明,但本发明完全不限定于这些实施例。本发明通过实施例进行了详尽的说明,但所属
技术领域:
的技术人员能够理解可以进行各种变更或修饰。因此,只要这样的变更或修饰不脱离本发明的范围,它们也包括在本发明的范围内。实施例中使用的各种试剂只要没有特别记载,则使用市售品。nmr谱使用al400(400mhz,日本电子(jeol))、mercury400(400mhz,agilentthechnologies)型光谱仪、或装备有400mnmr探头(protasis)的inova400(400mhz,agilentthechnologies))型光谱仪,在氘代溶剂中含有四甲基硅烷的情况下使用四甲基硅烷作为内标、在除此以外的情况下使用nmr溶剂作为内标进行测定,以ppm表示全部δ值。简写符号的含义如下所示。s:单峰d:双峰t:三重峰q:四重峰dd:双二重峰dt:双三重峰td:三重双峰tt:三重三重峰ddd:双双二重峰ddt:双双三重峰dtd:双三重双峰tdd:三重双二重峰m:多重峰br:宽峰brs:宽单峰粉末x射线衍射测定关于粉末x射线衍射,根据需要将适量的试验物质用玛瑙制研钵轻轻粉碎,之后按照下面的试验条件进行测定。装置:panalyticalempyrean靶:cux射线输出设定:40ma,45kv扫描范围:2.0~40.0°步长:0.026°发散狭缝:自动照射宽度:10.00mm试样宽度:10.00mm包括数据处理的装置的操作按照各装置中指示的方法和步骤进行。热分析测定(差示扫描量热测定(dsc测定))dsc测定按照下述试验条件进行测定。装置:tainstrumentsq1000试样:约1mg试样容器:铝制升温速度:以10℃/分钟升温至300℃氛围气体:氮氮气流量:50ml/min.包括数据处理的装置的操作按照各装置中指示的方法和步骤进行。比较例13-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的i型结晶的合成将按照国际公开第2012/093708号小册子和国际公开第2011/004610号小册子记载的制造方法得到的3-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的白色固体(3.58g)添加在乙醇(7.84ml)中,以室温搅拌2小时。过滤收集后用乙醇(7.84ml)清洗,之后以70~80℃减压干燥20小时,得到i型结晶(收量:2.40g、收率:61.2%、纯度:98.21%)。另外,i型结晶如图1所示在粉末x射线衍射图谱中显示衍射角(2θ)8.1°、10.9°、12.1°、14.0°、14.9°、16.2°、17.7°、20.2°、21.0°、21.5°、22.6°、24.3°、25.4°26.4°、27.0°、28.3°、30.2°、30.9°、31.5°、32.7°、34.7°、35.4°和36.6°的特征峰。1h-nmr(dmso-d6):δppm9.35(1h,d,j=4.88hz),8.93(1h,d,j=1.22hz),8.84(1h,brs),8.72(1h,d,j=1.95hz),8.70(1h,s),8.63(1h,d,j=1.22hz),8.60(1h,dd,j=8.29,1.95hz),8.46(1h,s),8.25(1h,d,j=8.29hz),8.22(1h,brs),8.12(1h,d,j=4.88hz),4.59(3h,s),3.95(1h,tt,j=6.83,6.83hz),3.21(2h,q,j=7.56hz),1.83(6h,d,j=6.83hz),1.75(3h,t,j=7.56hz):lrms(esi)m/z455[m+h]实施例13-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的ii型结晶的合成将按照国际公开第2012/093708号小册子和国际公开第2011/004610号小册子记载的制造方法得到的3-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的白色固体(4.0g)添加到丙酮(19.54ml)中,在加热回流下搅拌16小时。在放置冷却至室温后,将固体过滤收集,用丙酮(8.4ml)清洗后,以70~80℃进行16~24小时的减压干燥,得到ii型结晶(收量:1.59g,收率:57.0%、纯度98.37%)。另外,ii型结晶如图2所示在粉末x射线衍射图谱中显示衍射角(2θ)7.7°、8.0°、11.1°、12.5°、12.9°、14.2°、15.2°、15.8°、17.2°、17.7°、19.0°、20.2°、21.1°、22.5°、22.8°、23.5°、24.5°、26.1°、26.7°、27.4°、28.0°、28.7°、29.4°、30.0°、31.7°、35.1°、36.2°、36.9°和37.6°的特征峰。另外,如图3所示,差示扫描量热测定(dsc测定)的结果,ii型结晶在270℃附近显示吸热峰。1h-nmr(dmso-d6):δppm9.35(1h,d,j=4.88hz),8.93(1h,d,j=1.22hz),8.84(1h,brs),8.72(1h,d,j=1.95hz),8.70(1h,s),8.63(1h,d,j=1.22hz),8.60(1h,dd,j=8.29,1.95hz),8.46(1h,s),8.25(1h,d,j=8.29hz),8.22(1h,brs),8.12(1h,d,j=4.88hz),4.59(3h,s),3.95(1h,tt,j=6.83,6.83hz),3.21(2h,q,j=7.56hz),1.83(6h,d,j=6.83hz),1.75(3h,t,j=7.56hz):lrms(esi)m/z455[m+h]实施例23-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的ii型结晶的合成将按照国际公开第2012/093708号小册子和国际公开第2011/004610号小册子记载的制造方法得到的3-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的白色固体(400mg)添加到甲乙酮(2.8ml)中,在加热回流下搅拌16小时。放置冷却至室温后,将固体过滤收集,用甲乙酮(1.2ml)清洗后,以70~80℃进行16~24小时减压干燥,得到ii型结晶(收量:197mg、收率:60.9%、纯度98.83%)。实施例33-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的ii型结晶的合成将按照国际公开第2012/093708号小册子和国际公开第2011/004610号小册子记载的制造方法得到的3-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的白色固体(400mg)添加到乙腈(4.0ml)中,在加热回流下搅拌3小时。放置冷却至室温后,将固体过滤收集,用乙腈(1.2ml)清洗后,以70~80℃进行3小时减压干燥,得到ii型结晶(收量:120mg、收率:43.0%、纯度98.25%)。实施例43-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的ii型结晶的合成将按照国际公开第2012/093708号小册子和国际公开第2011/004610号小册子记载的制造方法得到的3-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的白色固体(400mg)添加到甲基异丁基酮(4.0ml)中,在加热回流下搅拌3小时。放置冷却至室温后,将固体过滤收集,用甲基异丁基酮(1.2ml)清洗后,以70~80℃进行3小时减压干燥,得到ii型结晶(收量:154mg、收率:55.3%、纯度96.89%)。实施例53-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的ii型结晶的合成将按照国际公开第2012/093708号小册子和国际公开第2011/004610号小册子记载的制造方法得到的3-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的白色固体(400mg)添加到2-丙醇(4.0ml)中,在加热回流下搅拌3小时。放置冷却至室温后,将固体过滤收集,用2-丙醇(1.2ml)清洗后,以70~80℃进行3小时减压干燥,得到ii型结晶(收量:108mg、收率:38.8%、纯度96.83%)。实施例63-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的ii型结晶的合成将按照国际公开第2012/093708号小册子和国际公开第2011/004610号小册子记载的制造方法得到的3-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的白色固体(400mg)添加到乙酸乙酯(4.0ml)中,在加热回流下搅拌3小时。放置冷却至室温后,将固体过滤收集,用乙酸乙酯(1.2ml)清洗后,以70~80℃进行3小时减压干燥,得到ii型结晶(收量:156mg、收率:56.0%、纯度96.45%)。实施例73-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的ii型结晶的合成将按照国际公开第2012/093708号小册子和国际公开第2011/004610号小册子记载的制造方法得到的3-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的白色固体(400mg)添加到乙酸丁酯(4.0ml)中,在加热回流下搅拌3小时。放置冷却至室温后,将固体过滤收集,用乙酸丁酯(1.2ml)清洗后,以70~80℃进行3小时减压干燥,得到ii型结晶(收量:164mg、收率:58.8%、纯度96.04%)。实施例83-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的ii型结晶的合成将按照国际公开第2012/093708号小册子和国际公开第2011/004610号小册子记载的制造方法得到的3-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的白色固体(400mg)添加到环戊基甲基醚(4.0ml)中,在加热回流下搅拌3小时。放置冷却至室温后,将固体过滤收集,用环戊基甲基醚(1.2ml)清洗后,以70~80℃进行3小时减压干燥,得到ii型结晶(收量:192mg、收率:68.8%、纯度95.68%)。实施例93-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的ii型结晶的合成将按照国际公开第2012/093708号小册子和国际公开第2011/004610号小册子记载的制造方法得到的3-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的白色固体(400mg)添加到乙酸丙酯(4.0ml)中,在加热回流下搅拌3小时。放置冷却至室温后,将固体过滤收集,用乙酸丙酯(1.2ml)清洗后,以70~80℃进行3小时减压干燥,得到ii型结晶(收量:172mg、收率:61.5%、纯度96.77%)。实施例103-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的ii型结晶的合成在按照国际公开第2012/093708号小册子和国际公开第2011/004610号小册子记载的制造方法得到的3-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺的白色固体(400mg)中加入乙酸甲酯(2.2ml),在加热回流下搅拌16小时。放置冷却至室温后,将固体过滤收集,用乙酸甲酯(0.94ml)清洗后,以80℃进行减压干燥,得到ii型结晶(收量:215.5mg、收率:68.6%、纯度98.06%)。试验例1血中浓度测定试验制造i型结晶和ii型结晶各自的给药液(50mg/10ml/kg)。使用口服给药用探头,对在摄食条件下饲养的小鼠(balb/ca)以每1kg体重10ml的容量口服给予这些给药液。给药后,使其回到小鼠用笼中,确认状态。笼内为可自由地摄取水和饲料的状态。在给药1、2、3、4、8和24小时后,用异氟烷将小鼠麻醉,使用毛细采血管从眼窝静脉丛采血60μl。采集的血液进行冰冷,通过离心操作分离血浆。使用lc-ms/ms,根据由多元反应监测(multiplereactionmonitoring)法测得的各血浆中的化合物1的浓度,使用pharsight公司制的软件phoenixwinnonlin(v6.3.0),利用对数线性梯形法算出auc0-24hr。将结果示于图4和表1。根据本试验可知,在auc0-24hr(给药后0~24小时的血中浓度-时间曲线下面积)中,ii型结晶显示i型结晶的约3倍的值。因此,本发明的ii型结晶的口服吸收性明显优异,作为口服用医药组合物有用。[表1]试验例2固体稳定性试验(加速试验)以下述条件测定将实施例1中得到的ii型结晶以40℃±2℃/75%rh±5%rh保存1个月、3个月和6个月时的固体稳定性。保存条件:40℃±2℃/75%rh±5%rh测定时间点:1个月、3个月和6个月保存量:6g保存容器:双重聚乙烯袋,捆扎带固定具+塑料桶试样溶液的制备方法:精密地称量ii型结晶100mg,添加乙腈·水混合液(4:1)将其溶解(不易溶解时照射超声波使其溶解),准确定容为200ml。准确称量该液体10ml,添加水·乙腈混合液(1:1),准确定容为20ml,制成试样溶液。hplc测定按照下述条件实施。柱:普通财团法人化学物质评价研究机构l-column2ods粒径:3μm,内径:4.6mm,长度:15cm测定波长:220nm流动相:流动相a:10mmol/l磷酸盐缓冲液(ph6.9),流动相b:乙腈流量:约1.0ml/分钟流动相的送液中,如下所述改变流动相a和b的混合比,控制浓度梯度。[表2]从注入后起的时间(分钟)流动相a(%)流动相b(%)0~25802025~4580→6520→3545~55653555~7065→1035→9070~80109080~80.110→8090→2080.1~958020将利用hplc分析测定并评价试样溶液中的类似物质量的结果示于表3。其中,类似物质是除3-乙基-4-{3-异丙基-4-(4-(1-甲基-1h-吡唑-4-基)-1h-咪唑-1-基)-1h-吡唑并[3,4-b]吡啶-1-基}苯甲酰胺以外检测到的物质。[表3]由该结果可知,化合物1的ii型结晶的类似物质的生成少,表现出优异的固体稳定性。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 含氮的稠环化合物以及使用其的有机发光器件的制造方法与工艺
- (杂)芳基1,3‑偶极化合物与(杂)环炔的环加成方法与制造工艺
- 一种具抗肿瘤活性的岩白菜素氮杂肉桂酸酯类化合物及其合成方法与制造工艺
- 一种含有氮杂环的化合物及其有机电致发光器件的制造方法与工艺
- 靛红杂合喹唑啉类化合物合成的靛红衍生物及其在制备抗肿瘤药物中的应用的制造方法与工艺
- 氮杂环类化合物及其应用的制造方法与工艺
- 含唑杂环的嘧啶类化合物,组合物及其用途的制造方法与工艺
- 一种具有抗肿瘤活性的高乌甲素氮杂肉桂酸杂合体及其合成方法与制造工艺
- 一种氮杂芳烃苄位与缺电子芳环偶联的方法与制造工艺
- 一种含氮杂环衍生物及使用该含氮杂环衍生物的有机发光器件的制造方法与工艺