合成咪唑稠杂环化合物的方法与流程
本发明涉及合成咪唑稠杂环化合物的方法,具体的,涉及采用氯化镨或其水合物、氯化钕或其水合物、氯化钐或其水合物、氯化铕或其水合物、纳米氧化锌、纳米氧化铈、纳米氧化钛作为催化剂合成咪唑稠杂环化合物的方法。
背景技术:
咪唑稠杂环化合物在医药、材料、农药和染料等领域有着极其广泛的应用,具有咪唑骨架的杂环化合物很多具有良好的生物活性,像上市药物如阿吡坦、佐利米定、奥普力农等等,因此咪唑稠杂环化合物的快速高效合成变得至关重要。
多组分反应是指三种或者三种以上的反应物发生一步化学反应,且生成的产物中含有所有起始原料的一种合成方法。多组分反应比传统的合成方法具有原子经济性高、反应时间短、条件温和、操作简单、收率高、污染小的优势,符合绿色化学的理念。gbb反应是一个四中心,三组分的多组分反应。该反应是由醛组分、胺组分和异腈组分在合适的催化剂、适当的温度条件下发生“一锅煮”反应,最终形成多取代咪唑稠杂环化合物。
gbb反应催化剂通常是酸,很少一部分是碱。已报到gbb反应催化剂主要有:
布朗斯特酸,如醋酸、高氯酸、纤维素硫酸、对甲苯磺酸、甲磺酸(参见[1]sharma,a.;li,h.y.synlett2011,10,1407.[2]huang,y.;hu,x.q.;shen,d.p;chen,y.f.;xu,p.f.mol.diversity.2007,11,73.[3]groebke,k.;weber,l.;mehlin,f.synlett1998,6,661.[4]che,c.;xiang,j.;wang,g.x.;fathi,r.;quan,j.m.;yang,z.j.comb.chem.2007,9,982.[5]mert-balci,f.;conrad,j.;meindl,k.;schulz,t.;stalke,d.;beifuss,u.synthesis.2008,22,3649.);
路易斯酸,如三氟甲基磺酸钪、氯化锌、氯化镧,七水合氯化镧、七水合氯化铈、氯化镁、氯化锆、氯化锡、氯化钌和氯化铟(参见[1]mckeown,m.r.;shaw,d.l.;fu,h.;liu,s.;xu,x.;marineau,j.j.;huang,y.;zhang,x.;buckley,d.l.;kadam,a.;zhang,z.;blacklow,s.c.;qi,j.;zhang,w.;bradner,j.e.j.med.chem.2014,57,9019.[2]shaabani,a.;soleimani,e.;sarvary,a.;rezayan,h.;maleki,a.chin.j.chem.2009,27,369.[3]xi,g.l.;liu,z.q.tetrahedron2015,71,9602.[4]shinde,a.h.;srilaxmi,m.;satpathi,b.;sharada,d.s.tetrahedronlett2014,55,5915.[5]guchhait,s.k.;madaan,c.tetrahedronlett.2011,52,56.[6]odell,lr.;nilsson,m.;gising,j.;lagerlund,o.;muthas,d.;nordqvist,a.;karlen,a.;larhed,m.bioorg.med.chem.lett.2009,19,4790.[7]guchhait,s.k.;maadan,c.synlett2009,4,628.[8]rousseau,a.l.;matlaba,p.;parkinson,c.j.tetrahedronlett.2007,48,4079.[9]rostamnia,s.;hassankhanib,a.rscadv.2013,3,18626.[10]akbarzadeh,r.;shakibaei,g.i.;bazgir,a.monatsh.chem.2010,141,1077.);
固体酸催化剂,如锰酸镧(参见sanaeishoara,t.;tavakkolia,h.;mohave,f.appl.catal.,a2014,470,56.);
离子液体,如1-丁基-3-甲基咪唑溴盐(参见shaabani,a.;soleimani,e.;maleki,a.tetrahedronlett.2006,47,3031.);
碱,如哌啶(参见lee,c.h.;hsu,w.s.;chen,c.h.;sun,c.m.eur.j.org.chem.2013,11,2201.)。
上述报道的催化剂存在很多问题,如反应时间长、反应条件苛刻、产量低、试剂昂贵、产生大量有毒的含金属废物等。
因此,需要发展新型、高催化活性、方便易得的催化剂。
技术实现要素:
为解决上述问题,发明人通过对gbb多组分反应催化剂的深入探索,提出如下技术方案:
本发明一方面涉及一种合成咪唑稠杂环化合物的方法,所述的反应如下式所示
其中
与n-h2和r1相连的环为含有至少一个n的五元或者六元杂芳环,或者为c6-c40的稠合杂芳环;所述与n-h2和r1相连的环被1-5个r1所取代,所述的r1为c1-c3的烷基、c1-c3的甲氧基、卤素、硝基、氰基;
r2选自:取代或未取代芳基、取代或未取代杂芳基、取代或未取代c1-c5的烷基或者c1-c3的甲氧基;优选的,所述取代基为c1-c5的烷基、卤素、硝基、氰基;
所述r3选自:c1-c6的烷基、c3-c7的环烷基、取代或者未取代的芳烷基、取代或者未取代的杂芳基烷基;优选的,所述取代基为c1-c5的烷基、卤素、硝基、氰基;
其特征在于,所述的催化剂含有氯化镨或其水合物、氯化钕或其水合物、氯化钐或其水合物、氯化铕或其水合物、纳米氧化锌、纳米氧化铈、纳米氧化钛中的至少一种。
优选的,所述的r2选自苯基、吖嗪、萘、甲基、乙基、正丙基、异丙基、甲氧基、乙氧基;任选地,所述的苯基、吖嗪、萘被1-3个选自甲基、乙基、正丙基、异丙基、甲氧基、乙氧基、卤素中的任意一个取代基所取代;
在本发明的一个优选实施方式中,所述的r3选自叔丁基、正丁基、环己基、甲基苯磺酰甲基、苄基、2-氯-6-苯甲基、苯乙基。
在本发明的一个优选实施方式中,所述的反应是在140℃以下进行,优选的,所述反应在惰性气氛保护下进行,所述惰性气氛优选为氮气和/或氩气。
在本发明的一个优选实施方式中,所述的反应溶剂为乙醇。
在本发明的一个优选实施方式中,所述的反应温度50-70℃,反应溶剂为乙醇,反应时间为0.5-6小时。在上述技术方案中,反应温度高可以加快反应;同时反应温度也可以在温和的条件下,比如室温条件下反应;从节约能源的角度考虑,优选的技术方案中,反应温度为40-70℃。
在本发明的一个另一个优选实施方式中,所述的反应温度为80-170℃,优选为120-140℃;在微波无溶剂条件下反应,并在10分钟以内完成反应。
在本发明的一个优选实施方式中,所述催化剂选自氯化镨或其水合物、氯化钕或其水合物、氯化钐或其水合物、氯化铕或其水合物中的至少一种,所述反应产率不低于95%。
在本发明的另一个优选实施方式中,所述的催化剂选自纳米氧化锌、纳米氧化铈、纳米氧化钛中的至少一种,优选的,所述的催化剂通过固液分离方式从反应体系中除去或者回收。
制备纳米氧化锌催化剂时,可以采用报道的合成方法(sadjadi,s,;eskandari,m.ultrasonicssonochemistry.2013,20,640.),其余催化剂市售购买获得。
在本发明的一个优选实施方式中,所述的反应完成之后冷却至室温终止反应,然后萃取反应液,萃取液用干燥剂干燥,过滤,然后减压除去溶剂,直接通过重结晶获得目标产物或者通过硅胶柱层析分离得到目标化合物。在上述技术方案中,优选的技术方案中萃取剂为二氯甲烷,干燥剂为无水硫酸钠,重结晶的溶剂为石油醚。在上述技术方案中,所述硅胶柱层析分离使用的洗脱剂为石油醚∶乙酸乙酯=4-6∶1的混合溶剂。
上述技术方案中,催化剂的量为原料摩尔数的1%以上,并且随着催化剂的量增大越有利反应的进行,而且在后续产物分离中,催化剂的用量不会有影响,因为氯化镨或其水合物、氯化钕或其水合物、氯化钐或其水合物、氯化铕或其水合物都是水溶性的,而目标产物在水中溶解度小,易溶于有机相,在萃取过程中容易实现分离,纳米氧化锌、纳米氧化铈、纳米氧化钛为不溶性固体能过通过过滤直接除去;优选的技术方案中,催化剂的用量为原料的摩尔数的2-12%,不仅高效催化,而且节约成本。
上述技术方案中,醛组分、胺组分和异腈组分的物质的量可不相等,优选的技术方案中,醛组分、胺组分和异腈组分的物质的量相等。
上述技术方案中,反应溶剂可以选择水、甲醇、乙醇、丙醇、甲苯、二氧六环、二氯甲烷、二甲基亚砜、聚乙二醇、二甲基甲酰胺和乙腈;同时反应也可以在无溶剂条件下反应,从反应产率高低的角度考略,优选的技术方案中,反应溶剂为乙醇。
由于上述技术方案的应用,本发明与现有技术相比具有至少具有下列优点中的一个或者多个或者全部:
本发明所使用的催化剂在提高反应活性的同时,缩短了反应时间,降低了催化剂的制备难度或购买成本,微波反应温度130℃,低于文献普遍报道的150℃-160℃,且且微波反应不需要使用溶剂,收率较高,纯化方法简单,大部分产物直接通过重结晶纯化,为gbb多组分反应提供多种可选择性。此外本发明的所使用的催化剂便宜易得、收率高、反应时间短、后处理简单,相对已有催化剂更有利于工业化制备咪唑稠杂环化合物。
附图说明
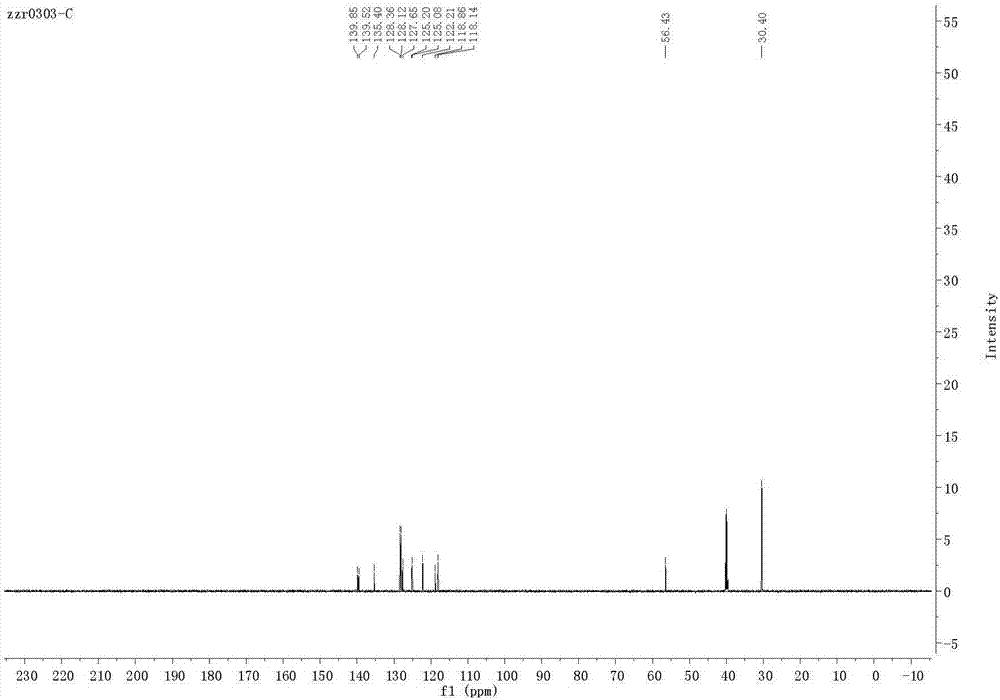
图1、图2、图3、图4分别为实施例一产物n-叔丁基-6-氯-2-苯基咪唑[1,2-a]吡啶-3-氨基的1hnmr、13cnmr图谱、高效液相谱图、高分辨质谱图。
具体实施方式
下面结合附图及实施例对本发明作进一步描述:
实施例一:无水氯化镨催化2-氨基-5-氯吡啶、苯甲醛和叔丁基异腈合成n-叔丁基-6-氯-2-苯基咪唑[1,2-a]吡啶-3-氨基(如下图)。
在10ml史莱克管中加入0.129克(1.00毫摩尔)2-氨基-5-氯吡啶、0.106克(1.00毫摩尔)苯甲醛、0.083克(1.00毫摩尔)叔丁基异腈、0.025克(10%毫摩尔)氯化镨和2ml乙醇,在氮气保护,温度60℃条件下,搅拌1小时,薄层色谱监测反应完全。冷却至室温,反应液转移至分液漏斗中,加入30ml蒸馏水和50ml二氯甲烷萃取分液,保留有机相,水相用二氯甲烷萃取两次,每次50ml,之后合并有机相,加入无水硫酸钠干燥过夜。抽滤,除去干燥剂,将滤液旋干,残余物用石油醚重结晶,真空干燥得到白色固体0.294克,产率98%,经高效液相测定纯度高于98%。
实施例二:六水氯化钕催化2-氨基-5-氯吡啶、苯甲醛和叔丁基异腈合成n-叔丁基-6-氯-2-苯基咪唑[1,2-a]吡啶-3-氨基。
在10ml史莱克管中加入0.129克(1.00毫摩尔)2-氨基-5-氯吡啶、0.106克(1.00毫摩尔)苯甲醛、0.083克(1.00毫摩尔)叔丁基异腈、0.036克(10%毫摩尔)六水氯化钕和2ml乙醇,在氮气保护,温度60℃条件下,搅拌1小时,薄层色谱监测反应完全。冷却至室温,反应液转移至分液漏斗中,加入30ml蒸馏水和50ml二氯甲烷萃取分液,保留有机相,水相用二氯甲烷萃取两次,每次50ml,之后合并有机相,加入无水硫酸钠干燥过夜。抽滤,除去干燥剂,将滤液旋干,残余物用石油醚重结晶,真空干燥得到白色固体0.288克,产率96%,经高效液相测定纯度高于97%。
实施例三:氯化钐催化2-氨基-5-氯吡啶、苯甲醛和叔丁基异腈合成n-叔丁基-6-氯-2-苯基咪唑[1,2-a]吡啶-3-氨基。
在10ml史莱克管中加入0.129克(1.00毫摩尔)2-氨基-5-氯吡啶、0.106克(1.00毫摩尔)苯甲醛、0.083克(1.00毫摩尔)叔丁基异腈、0.029克(10%毫摩尔)氯化钕在微波130℃条件下,反应10min,薄层色谱监测反应完全。冷却至室温,反应液转移至分液漏斗中,加入30ml蒸馏水和50ml二氯甲烷萃取分液,保留有机相,水相用二氯甲烷萃取两次,每次50ml,之后合并有机相,加入无水硫酸钠干燥过夜。抽滤,除去干燥剂,将滤液旋干,残余物用石油醚重结晶,真空干燥得到白色固体0.293克,产率98%,经高效液相测定纯度高于98%。
实施例四:六水氯化钐催化2-氨基-5-氯吡啶、苯甲醛和叔丁基异腈合成n-叔丁基-6-氯-2-苯基咪唑[1,2-a]吡啶-3-氨基。
在10ml史莱克管中加入0.129克(1.00毫摩尔)2-氨基-5-氯吡啶、0.106克(1.00毫摩尔)苯甲醛、0.083克(1.00毫摩尔)叔丁基异腈、0.036克(10%毫摩尔)六水氯化钐和2ml乙醇,在氮气保护,温度60℃条件下,搅拌1小时,薄层色谱监测反应完全。冷却至室温,反应液转移至分液漏斗中,加入30ml蒸馏水和50ml二氯甲烷萃取分液,保留有机相,水相用二氯甲烷萃取两次,每次50ml,之后合并有机相,加入无水硫酸钠干燥过夜。抽滤,除去干燥剂,将滤液旋干,残余物用石油醚重结晶,真空干燥得到白色固体0.291克,产率97%,经高效液相测定纯度高于98%。
实施例五:六水氯化铕催化2-氨基-5-氯吡啶、苯甲醛和叔丁基异腈合成n-叔丁基-6-氯-2-苯基咪唑[1,2-a]吡啶-3-氨基。
在10ml史莱克管中加入0.129克(1.00毫摩尔)2-氨基-5-氯吡啶、0.106克(1.00毫摩尔)苯甲醛、0.083克(1.00毫摩尔)叔丁基异腈、0.037克(10%毫摩尔)六水氯化铕和2ml乙醇,在氮气保护,温度60℃条件下,搅拌1小时,薄层色谱监测反应完全。冷却至室温,反应液转移至分液漏斗中,加入30ml蒸馏水和50ml二氯甲烷萃取分液,保留有机相,水相用二氯甲烷萃取两次,每次50ml,之后合并有机相,加入无水硫酸钠干燥过夜。抽滤,除去干燥剂,将滤液旋干,残余物用石油醚重结晶,真空干燥得到白色固体0.285克,产率95%,经高效液相测定纯度高于97%。
实施例六:纳米氧化锌催化2-氨基-5-氯吡啶、苯甲醛和叔丁基异腈合成n-叔丁基-6-氯-2-苯基咪唑[1,2-a]吡啶-3-氨基。
在10ml史莱克管中加入0.129克(1.00毫摩尔)2-氨基-5-氯吡啶、0.106克(1.00毫摩尔)苯甲醛、0.083克(1.00毫摩尔)叔丁基异腈、0.008克(10%毫摩尔)纳米氧化锌和2ml乙醇,在氮气保护,温度60℃条件下,搅拌1小时,薄层色谱监测反应完全。冷却至室温,反应液经过滤除去催化剂,反应液转移至分液漏斗中,加入30ml蒸馏水和50ml二氯甲烷萃取分液,保留有机相,水相用二氯甲烷萃取两次,每次50ml,之后合并有机相,加入无水硫酸钠干燥过夜。抽滤,除去干燥剂,将滤液旋干,残余物用石油醚重结晶,真空干燥得到白色固体0.276克,产率92%,经高效液相测定纯度高于96%。
实施例七:纳米氧化铈催化2-氨基-5-氯吡啶、苯甲醛和叔丁基异腈合成n-叔丁基-6-氯-2-苯基咪唑[1,2-a]吡啶-3-氨基。
在10ml史莱克管中加入0.129克(1.00毫摩尔)2-氨基-5-氯吡啶、0.106克(1.00毫摩尔)苯甲醛、0.083克(1.00毫摩尔)叔丁基异腈、0.017克(10%毫摩尔)纳米氧化铈和2ml乙醇,在氮气保护,温度60℃条件下,搅拌1小时,薄层色谱监测反应完全。冷却至室温,反应液经过滤除去催化剂,反应液转移至分液漏斗中,加入30ml蒸馏水和50ml二氯甲烷萃取分液,保留有机相,水相用二氯甲烷萃取两次,每次50ml,之后合并有机相,加入无水硫酸钠干燥过夜。抽滤,除去干燥剂,将滤液旋干,残余物用石油醚重结晶,真空干燥得到白色固体0.273克,产率91%,经高效液相测定纯度高于96%。
实施例八:纳米氧化钛催化2-氨基-5-氯吡啶、苯甲醛和叔丁基异腈合成n-叔丁基-6-氯-2-苯基咪唑[1,2-a]吡啶-3-氨基。
在10ml史莱克管中加入0.129克(1.00毫摩尔)2-氨基-5-氯吡啶、0.106克(1.00毫摩尔)苯甲醛、0.083克(1.00毫摩尔)叔丁基异腈、0.017克(10%毫摩尔)纳米氧化钛和2ml乙醇,在氮气保护,温度60℃条件下,搅拌1小时,薄层色谱监测反应完全。冷却至室温,反应液经过滤除去催化剂,反应液转移至分液漏斗中,加入30ml蒸馏水和50ml二氯甲烷萃取分液,保留有机相,水相用二氯甲烷萃取两次,每次50ml,之后合并有机相,加入无水硫酸钠干燥过夜。抽滤,除去干燥剂,将滤液旋干,残余物用石油醚重结晶,真空干燥得到白色固体0.270克,产率90%,经高效液相测定纯度高于96%。
产物n-叔丁基-6-氯-2-苯基咪唑[1,2-a]吡啶-3-氨基核磁氢谱见图1,核磁碳谱见图2,高效液相谱图见图三。核磁数据1hnmr(600mhz,dmso-d6)δ:8.50(s,1h),8.14(d,j=7.5hz,2h),7.51(d,j=9.4hz,1h),7.38(t,j=7.6hz,2h),7.27(t,j=7.3hz,1h),7.20(dd,j=9.4,1.5hz,1h),4.67(s,1h),0.98(s,9h);13cnmr(150mhz,dmso-d6)δ:139.85,139.52,135.40,128.36,128.12,127.65,125.14,122.21,118.86,118.14,56.43,30.40.hrmscalcdforc17h18cln3[m+h]+:300.1262,found300.1263。以上数据证明所得化合物为目标化合物。
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!