一种从莪术油中分离β‑榄香烯的方法与流程
(一)技术领域
本发明涉及一种从莪术油中分离β-榄香烯的方法,具体涉及一种利用高速逆流色谱(hsccc)从莪术油中分离β-榄香烯的方法。
(二)技术背景
β-榄香烯是存在于莪术油中的一种倍半萜类物质,是我国首先从姜科植物温莪术(拉丁学名:curcumaerhizoma)中提取的具有抗癌活性的天然抗癌药物。与传统抗癌药物比具有抗癌谱广、毒副作用小、疗效确切等优点。目前已广泛用于肝癌、肺癌、乳腺癌、宫颈癌、消化道肿瘤、膀胱癌、脑肿瘤以及其它浅表性肿瘤的临床治疗。β-榄香烯,英文名β-elemene,分子式c15h24,分子量204.355,分子结构如下:
莪术油作为中药莪术的主要活性成分,具有抗肿瘤、抗病毒、抗菌、抗炎、抗早孕、降酶等作用。β-榄香烯的分离一般采用减压精馏、硅胶层析,但这个方法需经过两次精馏以及后续经过柱层析除去杂质才能得到高纯度的β-榄香烯。逆流色谱是20世纪80年代发展起来的一种连续高效的液-液分配色谱分离技术,该技术可避免因不可逆吸附而引起的样品损失、失活、变性,与普通液相色谱相比其样品的制备量大、维护费用低,从而成为一种理想的制备分离手段。利用倍半萜类化合物可以和硝酸银形成π配位化合物的特性,通过在高速逆流色谱的流动相中加入银离子,经过一次逆流分离得到高纯度的β-榄香烯。
(三)
技术实现要素:
本发明目的是提供一种快速分离β-榄香烯的方法,本发明根据硝酸银与含有3个不饱和双键的β-榄香烯可以形成π配位化合物的特性,利用高速逆流色谱法快速地将β-榄香烯从莪术油中分离出来,具有更强的目的性,快捷高效。
本发明的技术方案如下:
一种从莪术油中分离β-榄香烯的方法,所述方法为:
将正己烷、甲醇、水按体积比2:0.3~1.7:1.7~0.3充分混合后静置,液体分层分别得到上相和下相;取下相添加硝酸银作为高速逆流色谱的固定相,上相作为流动相;称取莪术油,用上相和下相体积比1:1的混合液溶解后进样,经高速逆流色谱分离,紫外检测器检测,收集含β-榄香烯的流出液(tlc检测收集),蒸除溶剂并干燥,即得β-榄香烯产品(纯度在80%~99%)。
所述正己烷、甲醇、水的体积比优选2:1.5:0.5。
所述硝酸银的添加量以下相的体积计为0.05~0.3mol/l,优选0.1~0.25mol/l。
所述上相和下相体积比1:1的混合液的体积用量以莪术油的质量计为10~200ml/g,优选15~25ml/g。
具体的,所述高速逆流色谱分离的操作方法为:
先将固定相充满高速逆流色谱仪的多层线圈分离柱,设定高速逆流色谱仪柱温15~30℃(优选20℃),在500~1000r/min(优选800r/min)转速下,以0.5~5ml/min(优选2ml/min)的流速注入流动相,以波长200~300nm(优选210~280nm)的紫外检测器检测,当柱尾端有流动相流出时进样,收集含β-榄香烯的流出液(tlc检测收集),蒸除溶剂并干燥,即得β-榄香烯产品。
与现有技术比较,本发明的优势在于:本发明分离方法操作简便,高效快捷,溶剂消耗少;硝酸银对β-榄香烯具有π配位作用,可以选择性地从莪术油中分离出来,分离效果优异。
(四)附图说明
图1:实施例1的高速逆流色谱图;
图2:实施例2的高速逆流色谱图;
图3:实施例3的高速逆流色谱图;
图4:逆流色谱2号峰的gc-ms图谱;
图5:逆流色谱2号峰的质谱图。
(五)具体实施方式
下面以具体实施例来对本发明的技术方案作进一步说明,但本发明的保护范围不限于此。
下述实施例中,采用的高速逆流色谱型号为:hsccc-tbe200v
实施例1:
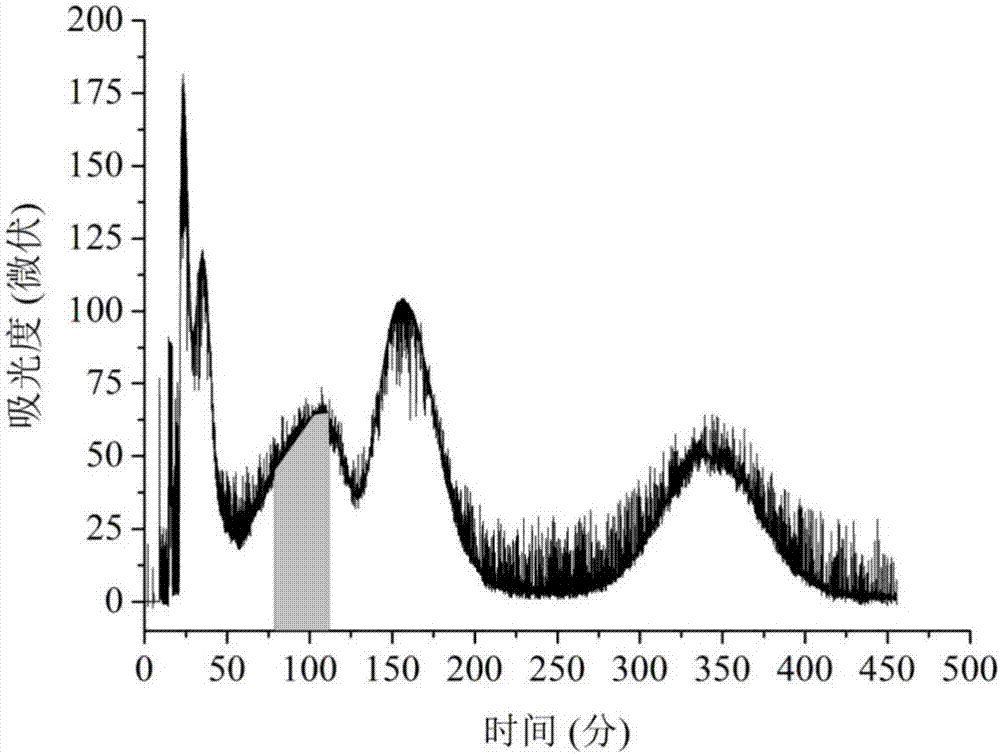
按照溶剂体系正己烷-甲醇-水(2:1.5:0.5,v/v/v)在分液漏斗中混合,充分振荡,静止,临用前分层,在下相中加入0.2mol/l硝酸银选为固定相,上相选为流动相;柱温设定为20℃,先用恒流泵将溶剂体系的下相打入hsccc的聚四氟乙烯螺旋管,直至注满整个管子,然后开启hsccc,逆时针旋转,转速为802.5prm,同时将流动相以2.0ml/min流速从柱的首端泵入hsccc分离柱;等到柱尾端流出流动相时,表明hsccc中两相已达到平衡,通过六通阀将样品注入hsccc(502.3mg莪术油样品溶解于8ml等体积混合的上下相溶剂中);以波长254nm的紫外检测器检测流出液,见图1,图1中阴影部分为β-榄香烯,收集含β-榄香烯的流出液(tlc检测收集),蒸除溶剂并干燥,得β-榄香烯28.3mg,纯度为95%。
实施例2:
按照溶剂体系正己烷-甲醇-水(2:1.5:0.5,v/v/v)在分液漏斗中混合,充分振荡,静止,临用前分层,在下相中加入0.15mol/l硝酸银选为固定相,上相选为流动相;柱温设定为20℃,先用恒流泵将溶剂体系的下相打入hsccc的聚四氟乙烯螺旋管,直至注满整个管子,然后开启hsccc,逆时针旋转,转速为800.4prm,同时将流动相以2.0ml/min流速从柱的首端泵入hsccc分离柱;等到柱尾端流出流动相时,表明hsccc中两相已达到平衡,通过六通阀将样品注入hsccc(440.3mg莪术油样品溶解于8ml等体积混合的上下相溶剂中);以波长254nm的紫外检测器检测流出液,见图2,图2中阴影部分为β-榄香烯,收集含β-榄香烯的流出液(tlc检测收集),蒸除溶剂并干燥,将对应峰的洗脱液的溶剂挥干,得β-榄香烯21.5mg,纯度为96%。
实施例3:
按照溶剂体系正己烷-甲醇-水(2:1.5:0.5,v/v/v)在分液漏斗中混合,充分振荡,静止,临用前分层,在下相中加入0.15mol/l硝酸银选为固定相,上相选为流动相;柱温设定为20℃,先用恒流泵将溶剂体系的下相打入hsccc的聚四氟乙烯螺旋管,直至注满整个管子,然后开启hsccc,逆时针旋转,转速为800.4prm,同时将流动相以2.0ml/min流速从柱的首端泵入hsccc分离柱;等到柱尾端流出流动相时,表明hsccc中两相已达到平衡,通过六通阀将样品注入hsccc(545.4mg过柱后莪术油样品溶解于8ml等体积混合的上下相溶剂中);以波长254nm的紫外检测器检测流出液,见图3,图3中阴影部分为β-榄香烯,收集含β-榄香烯的流出液(tlc检测收集),蒸除溶剂并干燥,得β-榄香烯147.6mg,纯度为99%。所得β-榄香烯产品的gc-ms图见图4、图5,图4为β-榄香烯的总离子流图,图5为质谱图。
对比例
采用真空间歇精馏方法对从莪术精油提取榄香烯的精制过程进行了研究,得到适宜的工艺条件。需采用二次精馏,一次精馏回流比1.0~3.0,收集53~63℃馏分,作为二次精馏原料。二次精馏回流比3.0~7.0,收集55~58℃馏分。产品榄香烯的质量分数为88.0%,收率为66.0%。
通过对比可知,以硝酸银作为选择性试剂采用高速逆流色谱法从莪术油中分离得到的β-榄香烯纯度更高,操作时间更短。
- 还没有人留言评论。精彩留言会获得点赞!