一株可以高效分泌纳豆激酶的枯草芽孢杆菌株及高纯纳豆激酶制备工艺的制作方法
本发明涉及一种微生物菌株及其发酵制备高纯酶制剂的工艺,特别涉及一种高效分泌纳豆激酶的枯草芽孢杆菌株及制备高纯纳豆激酶酶制剂的工艺。
背景技术:
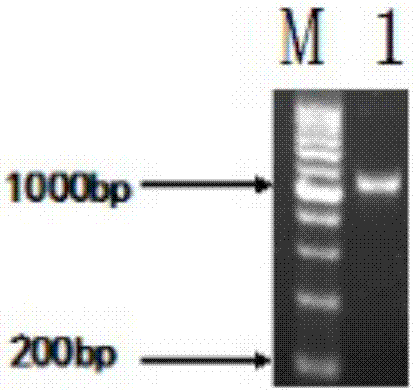
:纳豆是黄豆经过纳豆枯草杆芽孢杆菌发酵制成的发酵豆制品食品。纳豆激酶(nk)是一种由纳豆枯草芽孢杆菌产生的丝氨酸蛋白酶,存在于纳豆中。nk除在体内外有强烈的纤维蛋白溶解活性外,还有促进血液流动、防血小板凝聚和降血压等功效。nk于1980年首次由须见洋行博士发现,其相对分子质量(mr)为27000,等电点为8.6±0.3。由血栓引起的心血管疾病严重危害人类健康,目前溶栓剂包括注射类降解药物(尿激酶、链激酶和组织型纤维蛋白原激活剂)和类组织纤维蛋白酶的蛋白(纳豆激酶和蚓激酶)两大类。注射类降解药物高度依赖体内组织纤维蛋白酶原的水平,存在一定毒性,半衰期短且成本高。而nk属于天然发酵产物,安全性高,具有体内作用时间长、价格低廉、有预防作用及易于规模化生产等优点,已发展为保健食品,其临床应用已得到美国fda认可。提高nk工业化生产的产量和纯度,对治疗血栓疾病有重要意义。采用优化启动子和信号肽、选用蛋白酶缺失的菌株以降低nk降解或者直接改造nk基因序列等技术手段以达到提高nk产量的目的。nk在枯草芽孢杆菌中可实现可溶性表达,wus等构建了枯草芽孢杆菌工程菌,并通过优化启动子使nk的表达量增加了136%,达1999u/ml(平板法)。其他细菌,例如大肠杆菌、地衣芽孢杆菌、乳酸链球菌和酵母菌等也通过基因工程发酵nk。但这些技术也存在一些需要解决的问题,如在大肠杆菌中表达出没有活性的包涵体;在其他工程菌中nk虽然能够可溶性表达,但酶产量低,纯化回收过程复杂,有待进一步改进工程菌构造。nishitoy等人已经证明,纳豆发酵菌属于枯草芽孢杆菌的一个亚种。枯草芽孢杆菌无致病性、无明显的密码子偏好性,具备包含转录、翻译、蛋白质折叠和分泌等机制,常常作为工程菌株构建载体。本实验前期验证b.subtilis168含有nk基因序列,但不能表达有活性的nk。采用分子生物学方法,依据b.subtilis168的基因密码子偏好性,优化nk-aprn基因编码前30个氨基酸的序列,并加入六个组氨酸标记,以pht01为表达载体,成功构建工程菌,优化发酵条件后最高酶活可达289u/ml,高密度发酵发酵液中最高酶活达到7778u/ml,经亲和柱层析制备高纯纳豆激酶酶制剂最高酶活为981731u/g,为纳豆激酶基因工程进一步研究和纳豆激酶的工业化生产奠定基础。技术实现要素:本发明的目的在于克服天然菌种发酵性能的不足,提供一株可以高效分泌纳豆激酶的枯草芽孢杆菌工程菌株和工程菌株用于发酵制备高纯纳豆激酶酶制剂的工艺。本研究利用pcr技术扩增纳豆枯草芽孢杆菌的aprn基因,并依据枯草芽孢杆菌的密码子偏爱性优化了起始30个氨基酸的密码子,构建了重组表达质粒pht01-aprn,经限制性酶切、pcr扩增和测序验证了其正确性。通过电击法将pht01-aprn导入枯草芽孢杆菌168(bacillussubtilis168),利用氯霉素抗性筛选获得工程菌。经iptg诱导表达,摇瓶发酵培养最高酶活为289u/ml,高密度发酵发酵液中最高酶活达到7778u/ml,经亲和柱层析制备高纯纳豆激酶酶制剂最高酶活为981731u/g。为解决上述问题,本发明采用以下技术方案:一株可以高效分泌纳豆激酶的枯草芽孢杆菌工程菌株,其特征在于,所述菌株的分类命名为:枯草芽孢杆菌(bacillussubtilis),保藏编号为cgmccno:13496,保藏单位为中国微生物菌种保藏管理委员会普通微生物中心(cgmcc),保藏地址为北京市朝阳区北辰西路1号院3号,保藏日期为2016年12月26日。本发明公开了构造高效分泌纳豆激酶的枯草芽孢杆菌工程菌株的方法、摇瓶培养发酵产酶条件以及酶活测定方法,操作如下:步骤一:nk-aprn基因片段的扩增及优化以b.subtilis168菌液为第一次扩增的模版,以表1所列f1-r1引物对为第一次扩增引物,凝胶电泳验证;胶回收产物为第二次pcr扩增模版,以表1所列f2-r1引物对为第二次扩增引物,凝胶电泳验证;第二次胶回收产物为第三次pcr扩增模版,以表1所列f3-r2his引物对为第三次扩增引物,扩增出优化了前30个密码子的aprn基因片段。步骤二:目的基因克隆步骤一扩增出的nk基因片段与pmd19-t载体连接,将连接产物采用热击方法转化到e.colidh5α感受态中,涂布amp抗性平板。步骤三:重组质粒的构建及验证质粒pht01和提取步骤二中阳性菌的克隆质粒双酶切,用胶回收试剂盒回收纯化目的片段,t4连接酶低温过夜酶连,产物转化e.colidh5α感受态细胞。菌落pcr鉴定后,取正确的阳性克隆活化培养,提取质粒测序。步骤四:工程菌的构建及鉴定制备b.subtilis168的感受态细胞,将步骤四中提取的质粒电转感受态细胞。随机挑取氯霉素抗性平板上长出的5-8个单菌落,菌落pcr鉴定,取正确的阳性克隆活化培养,提取质粒测序。步骤五:工程菌发酵活化工程菌培养种子液,以氯霉素作为抗生素,种子液转接发酵培养基培养,od600在0.8-1.0降温至25℃至30℃,加入终浓度为0.2-1.0mm的iptg诱导培养。步骤六:纳豆激酶酶活的测定本发明采用四肽底物法检测纳豆激酶酶活。制作对硝基苯胺的标准曲线。nk活性单位被定义为nk每分钟释放1nmol硝基苯胺的含量。本发明还公开了工程菌高密度发酵的发酵产酶条件和高纯纳豆激酶酶制剂的制备工艺,操作如下:(1)高密度发酵接种:活化菌种,在37℃、200rpm培养条件下制备一级种子液和二级种子液,种龄为12h的二级种子液按照10%接种量接种至发酵培养基(氯霉素终浓度为5μg/ml、消泡剂0.01%)。发酵培养基(1l):葡萄糖28.5g,甘油57g,酵母提取物60g、吡啶二羧酸0.167g、nh4cl3g、k2hpo4·3h2o1g、mnso4·h2o0.032g、feso4·7h2o0.05g、cocl2·6h2o0.01g、zncl20.01g、mgso4·7h2o2g、cacl2·2h2o5g,补水至1000ml。发酵:搅拌速率初始设置为800rpm,通气量为12l/min。设置参数,控制罐内温度为37℃、溶氧量为30%、发酵液的ph7.0,依据溶氧情况调整搅拌速率和通气量。蛋白诱导表达:取样检测od600,至对数前期降温至28℃,加入iptg开始诱导发酵。补料:补料培养基(1l):甘油250g,酵母提取物100g,补水至1000ml。检测发酵液od600和发酵培养基中甘油的消耗情况,分别采用一次补料和连续补料方式补料培养。终止发酵:发酵培养24-28h后,终止发酵。(2)高纯纳豆激酶酶制剂的制备发酵液经冷冻离心后收集上清液、(nh4)2so4沉淀去除杂蛋白并沉淀nk、镍柱纯化、脱盐并浓缩、加冷冻保护剂和冷冻干燥等步骤,制成高纯度纳豆激酶酶制剂,纳豆激酶酶活达到981713u/g。本发明具有如下的有益效果:构造枯草芽孢杆菌的纳豆激酶工程菌,实现纳豆激酶在其胞外表达,经优化发酵培养条件,酶活高达289u/ml,可进一步用于工业化液体发酵生产。附图说明表1nk-aprn基因pcr扩增引物。表2nk-aprn基因pcr扩增体系。图1琼脂糖凝胶电泳鉴定nk-aprn基因pcr扩增电泳图。图2琼脂糖凝胶电泳鉴定aprn基因克隆pcr扩增电泳图。图3琼脂糖凝胶电泳鉴定pmd19-t:aprn质粒酶切电泳图。图4琼脂糖凝胶电泳鉴定pht01质粒酶切电泳图。图5琼脂糖凝胶电泳鉴定pht01:aprn质粒构建电泳图。图6琼脂糖凝胶电泳鉴定工程菌构建电泳图。图7对硝基苯胺标准曲线。图8工程菌酶活和细胞生长图。图9nk高密度发酵od600、酶活。具体实施方式以下对本发明的实施例作详细说明:本实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和过程,但本发明的保护范围不限于下述的实施例。下列实施例中未注明具体条件的实验方法,通常按照常规条件,例如sambrook等分子克隆:实验室手册(newyork:coldspringharborlaboratorypress,1989)中所述的条件,或按照制造厂商所建议的条件。实施例1步骤一:nk-aprn基因片段的扩增及优化lb液体培养基:10g/l蛋白胨、10g/l氯化钠、5g/l酵母提取物。lb固体培养基:lb液体培养基、15g/l琼脂糖。amp溶液:100mg/ml。根据genbank收录纳豆激酶基因序列(gi:608796180),依据宿主菌b.subtitle密码子偏好(http://www.kazusa.or.jp/codon/cgi-bin/showcodon.cgi?species=1423),利用生物软件oligo设计引物(表1),扩增aprn基因并优化它的前30个氨基酸密码子。引物由生工生物工程有限公司合成。将保藏的b.subtilis168菌株活化后,按照2%的接种量接种于lb培养基中,37℃、200rpm培养12h。目的基因的克隆采用over-lappingpcr的方法,即以菌液为第一次pcr(德国eppendrof公司)扩增模版,将凝胶电泳验证后的胶回收(德国eppendrof公司)产物作为第二次pcr扩增模版,再以此次胶回收产物为模版进行第三次pcr扩增。引物依次为f1-r1、f2-r1和f3-r2his,扩增并优化源于b.subtilisnatto的aprn基因,如图1。扩增引物如表1,组合引物依次为引物依次为f1-r1、f2-r1和f3-r2his,扩增体系如表2,扩增反应条件:94℃预变性10min,94℃变性40s,54℃退火40s,72℃延伸30s,共35个循环,72℃延伸10min。步骤二:目的基因克隆基因克隆方法参照《分子克隆实验指南》。试剂盒回收并纯化第三次pcr扩增产物,产物通过ta克隆连到质粒pmd19-t。pmd19-t载体连接体系:dna4.5μl,pmd19-t0.5μl,缓冲溶液i5μl,合计10μl,4℃过夜连接。通过热激转化方法将连接产物导入大肠杆菌感受态中,涂布amp抗性平板(终浓度100μg/ml)。提取阳性克隆菌落质粒dna,通过限制性内切酶酶切图谱分析和pcr产物测序验证,将正确的携带pmd19-t:aprn的质粒菌株置于甘油管-80℃保存。步骤三:重组质粒的构建及工程菌构造pmd19-t:aprn由限制性内切酶ecorv和bamhi酶切,切下的aprn基因片段插入pht01的smaⅰ和bamhi酶切位点之间,通过热激转化方法将连接产物导入大肠杆菌感受态中,涂布amp抗性平板(终浓度100μg/ml)。提取阳性克隆菌落质粒dna,通过限制性内切酶酶切图谱分析和pcr产物测序验证,将正确的携带pht01:aprn的质粒菌株置于甘油管-80℃保存。步骤四:工程菌的构建及鉴定b.subtilis168的感受态细胞制备和电击转化参考xue的方法。随机挑取氯霉素抗性平板上长出的单菌落,提取阳性克隆菌落质粒dna,通过限制性内切酶酶切图谱分析和pcr产物测序验证,将正确的携带pht01:aprn的质粒菌株置于甘油管-80℃保存。步骤五:纳豆激酶工程菌发酵:发酵培养基:20g/l葡萄糖、20g/l酵母提取物、2g/lk2hpo4·3h2o、1g/lmgso4·7h2o,2%甘油和1mm吡啶二羧酸,ph调至7.0-8.0。取甘油管中工程菌以2%接种量接种于lb培养基(含5μg/ml氯霉素)中,200rpm、37℃过夜培养。以2%接种量将种子液接种于25ml发酵培养基a中,37℃、200rpm摇床培养,至od600在0.8-1.0之间,降温至25℃。接入终浓度为0.5mm的诱导剂iptg,在不同发酵时间测定菌液的od600和nk酶活,如图8。步骤六:四肽底物法检测酶活首先制作对硝基苯胺的标准曲线:配制对硝基苯胺样品溶液,浓度分别为0.02、0.03、0.04、0.06和0.08mmol,在405nm下测定不同浓度硝基苯胺样品溶液的吸光值,以水作为空白对照。发酵液4℃、8000r/min离心1min,将上清稀释成不同浓度作为检测样品液。检测方法在文献基础上加以改进:取100μl样品稀释液与50μl终浓度为0.5mm的四肽底物(d-val-leu-lys-对硝基苯胺)混合并在37℃水浴下反应1min,加入75μl50%的冰醋酸终止反应,在405nm下测定反应液吸光值。nk活性单位被定义为nk每分钟释放1nmol硝基苯胺的含量。实施例2接种:活化菌种,在37℃、200rpm培养条件下制备一级种子液和二级种子液,种龄为12h的二级种子液按照10%接种量接种至发酵培养基(氯霉素终浓度为5μg/ml、消泡剂0.01%)。发酵培养基(1l):葡萄糖28.5g,甘油57g,酵母提取物60g、吡啶二羧酸0.167g、nh4cl3g、k2hpo4·3h2o1g、mnso4·h2o0.032g、feso4·7h2o0.05g、cocl2·6h2o0.01g、zncl20.01g、mgso4·7h2o2g、cacl2·2h2o5g,补水至1000ml。发酵:搅拌速率初始设置为800rpm,通气量为12l/min。设置参数,控制罐内温度为37℃、溶氧量为30%、发酵液的ph7.0,依据溶氧情况调整搅拌速率和通气量。蛋白诱导表达:检测od600,当od600等于10时降温至28℃加入终浓度为0.2-1.0mmiptg进行蛋白质诱导表达。补料:补料培养基(1l):甘油250g,酵母提取物100g,补水至1000ml。连续补料培养,设置补料程序为:发酵3h,od600=10开始补料,11-14h补料速率为250ml/h补料4h,15-18h再以170ml/h的速率补料4h,19-20h最后以170ml/h的速率补料2h。终止发酵:发酵培养24h后,终止发酵。经检测,发酵20h酶活可达到7778.02iu/ml(rsd%=0.8),od600为173.5±0.85,见图9。实施例3发酵液经4℃、9000r/m冷冻离心10min,取上清液;将研细的(nh4)2so4按照20%的饱和度加入发酵上清液中,4℃下保存过夜,10000r/m离心30min,弃去沉淀;继续向发酵上清液中加硫酸铵至60%饱和度,4℃下保存过夜,10000r/m离心30min,弃去上清液,回收率达到72%。实施例4平衡镍柱:吸取1ml含镍填料至纯化柱中,先用8倍体积去离子水清洗2遍,再用8倍体积结合液清洗三次。孵育:结合液将近流尽时,加入60ml发酵上清液,至4℃垂直旋转冰箱孵育4h,带有histag标签的nk与填料结合。漂洗:下端排尽未与镍柱结合的液体,加入10倍柱体积的漂洗液洗杂蛋白,在冰上垂直反转5min,重复4次。洗脱:用移液枪吸取1.5ml洗脱液垂直摄入填料正中间将填料充分垂悬,冰上静置2min,收集流出液。重复洗脱10次左右。洗脱液采用5kd的超滤管,300g离心,以纯水作为稀释液,以达到浓缩纳豆激酶和除盐的目的。向浓缩纳豆激酶溶液中加入一定浓度冷冻保护剂,冷冻干燥即制得高纯度纳豆激酶酶制剂,经检测纳豆激酶酶活达到981713u/g。表1nk-aprn基因pcr扩增引物注:加下划线和波浪线部分表扩增重复;黑体加粗并加下划线部分表示酶切位点;斜体加下划线部分表示加入六个组氨酸。表2nk-aprn基因pcr扩增体系组分体积(μl)基因组dna模板2引物-f1引物-r1dntp210×buffer2.5taqdna聚合酶0.25h2o16.25total25当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1