一种荧光化合物及其生产方法与流程
本发明属于化学领域,具体是2,6-二溴-9,10-双[2-[三(1-甲基乙基)甲硅烷基乙炔]蒽的生产方法。以及以2,6-二溴-9,10-双[2-[三(1-甲基乙基)甲硅烷基乙炔]蒽作为有效成分的荧光物质。
背景技术:
物质分子或原子在一定条件下吸收辐射能而被激发到较高电子能态后,在返回基态的过程中将以不同的方式释放能量,其发射光既是荧光。
荧光物质有许多实际应用领域,包括矿物学、宝石学、医药、化学传感器(荧光光谱)、荧光标记、染料、生物探测器、宇宙射线检测,最常用的有荧光灯。并在近年来在化学领域得到了广泛的应用。如荧光指示牌、荧光棒等。
通过化学合成的方法制备荧光化合物,其目的在于可以通过以结构较简单的化合物为原料,经过一系列反应过程制得具有复杂成分的目标物质。
荧光的产生与化合物分子结构的具有如下关系:
1.跃迁类型:
π*-π跃迁的荧光效率高,系间跨越过程的速率常数小,有利于荧光的产生。
2.共轭效应:
提高共轭度有利于增加荧光效率并产生红移。
3.刚性平面结构:
可降低分子振动,减少与溶剂的相互作用,故具有很强的荧光。
4.取代基效应:
芳环上有供电基,使荧光增强。
本发明者为寻找一种荧光效果更好的荧光材料,使符合上述有利于荧光产生的分子结构,设计一种生产该化合物的方法。利用化学合成的方法,设计出一种生产该荧光化合物的方法,并对该化合物进行提纯、鉴定与紫外荧光效率的检测。
技术实现要素:
本发明的目的在于提供一种2,6-二溴-9,10-双[2-[三(1-甲基乙基)甲硅烷基乙炔]蒽的生产方法。从而开发出一种具有较好荧光特性的化合物。
为实现上述目的,本发明提供如下技术方案:
作为本发明的第一步反应:
(1)以使用2,6-氨基蒽醌为原料,通过t-buono催化进行桑德迈尔反应,使氨基由溴基取代;
(2)将反应所得物质加入35%盐酸(hcl)溶液,并使用ch3cn清洗将所得产物重结晶;
(3)将所得重结晶产物过滤并获得土黄色粉末状即2,6-溴基蒽醌。
作为本发明第二步反应:
使用丁基锂作为引发剂的阴离子反应,将三异丙基硅基乙炔加成在2,6-溴基蒽醌的9,10位上,并形成羟基;
使用氯化亚锡将形成的羟基切除,得到2,6-二溴-9,10-双[2-[三(1-甲基乙基)甲硅烷基乙炔]蒽;
使用硅胶色谱柱进行提纯,分别为200-300目与40-63目各进行一次;
收集所得产物,使用旋转蒸发仪去除溶剂,即得到2,6-二溴-9,10-双[2-[三(1-甲基乙基)甲硅烷基乙炔]蒽。
与现有技术相比,本发明的有益效果是:
本发明的荧光化合物符合具有较好荧光效果的分子结构设计。本发明的生产制作方法简单,成本低廉,操作工艺简单;本发明的荧光化合物,荧光效果好,熔点高,稳定性好,可长期室温保存;采用的合成工艺路线清晰,可重现性高,生产过程节约试剂,具有较高的工业生产与实际应用价值。
附图说明
下面结合附图对本发明作进一步说明:
图1本发明中涉及的桑德迈尔反应机理,图中所示为一侧反应,对侧相同;
图2本发明中涉及的阴离子反应机理,图中所示为一侧反应,对侧相同;
图3本发明中第一步反应产物2,6-溴基蒽醌的1h-nmr图谱;
图4本发明中第一步反应产物2,6-溴基蒽醌的13c-nmr图谱;
图5本发明中第一步反应产物2,6-溴基蒽醌的ei质谱;
图6本发明中第一步反应产物2,6-溴基蒽醌的ftir红外图谱;
图7本发明中第二步反应所得荧光化合物6-二溴-9,10-双[2-[三(1-甲基乙基)甲硅烷基乙炔]蒽的ftir红外图谱;
图8本发明中第二步反应所得荧光化合物6-二溴-9,10-双[2-[三(1-甲基乙基)甲硅烷基乙炔]蒽的ei质谱;
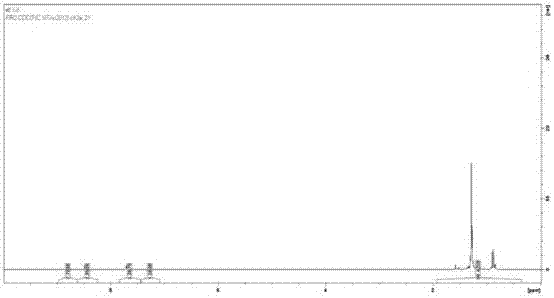
图9a本发明中第二步反应所得荧光化合物6-二溴-9,10-双[2-[三(1-甲基乙基)甲硅烷基乙炔]蒽的1h-nmr图谱;
图9b本发明中第二步反应所得荧光化合物6-二溴-9,10-双[2-[三(1-甲基乙基)甲硅烷基乙炔]蒽的1h-nmr图谱的芳香区放大图;
图10本发明中第二步反应所得荧光化合物6-二溴-9,10-双[2-[三(1-甲基乙基)甲硅烷基乙炔]蒽的13c-nmr图谱;
图11本发明中第二步反应所得荧光化合物6-二溴-9,10-双[2-[三(1-甲基乙基)甲硅烷基乙炔]蒽的紫外可见(uv-vis)光谱图。
具体实施方式
下面结合具体实施方式对本专利的技术方案作进一步详细地说明。如无特殊说明,均为常规方法;所用的实验材料,如无特殊说明,均为自常规生化试剂厂商购买得到的。
作为本发明的第一步反应:
使用氨基蒽醌10g,t-buono10g,cubr222g,及ch3cn170ml加入500ml一口圆底烧瓶,磁力搅拌并氮气保护,搅拌并加热至65℃持续2小时,再接着加入35%盐酸(hcl)溶液100ml至烧瓶。所得产物过滤并用ch3cn清洗并将所得产物重结晶,方法是采用1,4-二氧六环(1,4-dioxane)500ml加入500ml一口圆底烧瓶,磁力搅拌并使用氮气保护,加热至115℃持续40分钟,溶液再次过滤并获得土黄色粉末状产物10.46g。使用薄层色谱(tlc)检测,使用硅胶60f524(merck公司,0.3mm),质谱(ei);(m/z):364,366,368(m+).mp=283-284c如图5所示。核磁1hnmr(400mhz,cdcl3)δh/ppm;8.46(d,j=1.99hz,2h),8.20(d,j=8.31hz,2h),7.97(dd,j=8.31,2.02hz,2h)如图3所示。核磁13c-nmr(400mhz,cdcl3)δc/ppm:137.41,130.38,129.16如图4所示。使用的核磁仪器为brukerav250。图6的红外图谱证明了2,6位的氨基已经被溴基取代,使用仪器为perkinelemerspectrum65,该反应机理为桑德迈尔(sandmeyer)反应如图1所示。本步骤产率为35.8%,反应产物熔点为287.1℃。
作为本发明第二步反应:
将第一步所得产物进行下一步反应,第二步反应可以分为2个阶段,包括首先将四氢呋喃(thf)20ml与6.75ml的三异丙基硅基乙炔(triisopropylsilylacetylene)(30.1mmol)加入预先干燥过的圆底烧瓶500ml并加入磁力搅拌器,冰浴至0℃并氮气保护,接着加入20.5ml丁基锂(n-buli)(32.8mmol,1.6m溶于戊烷)混合物,并搅拌至少2小时,接着加入预先溶于150ml的四氢呋喃中的5g2,6-二溴-9,10-蒽醌(2,6-dibromo-9,10-anthraquinone)(13.7mmol),并氮气保护。接着混合物将于室温下至少搅拌24小时,将水加入反应体系终止反应,接着将混合物产物倒入饱和氯化铵中。使用乙酸乙酯ea(ethylacetate)来提取混合物。使用分液漏斗将有机层分离,所得分液层使用卤水(饱和氯化钠)清洗并使用无水硫酸镁(anhydrousmgso4)干燥。第二个阶段是首先蒸发溶液将所得有机产物再次溶解于150ml的四氢呋喃中并使用氮气保护,接着将氯化亚锡(sncl2·2h2o)(15g,66.5mmol,溶于70ml50%乙酸)以逐步滴加的方式加入反应体系,反应体系在室温下搅拌12小时,然后将其倒入水中并用乙酸乙酯(ea)提取。使用分液漏斗将有机层分离并使用碳酸氢钠(nahco3)与卤水(饱和氯化钠)清洗,并用无水硫酸镁(anhydrousmgso4)干燥。产物使用旋转蒸发仪去除溶剂,并2次使用硅胶色谱柱进行提纯,使用(正)己烷(hexane)作为洗脱剂,将通过薄层色谱法(tlc)测试过的产物收集并合并,使用旋转蒸发仪去除(正)己烷,得到产物2,6-二溴-9,10-双[2-[三(1-甲基乙基)甲硅烷基乙炔]蒽(3.7g)。如图9a与9b所示1hnmr(cdcl3,ppm)δ=8.79(d,2h),8.43(d,2h),7.65(dd,2h),1.25(m,42h)。如图10所示13c-nmr(cdcl3,ppm)δ=133.23,130.95,130.89,129.49,128.96,121.99,118.16,106.31,102.18,18.83,11.46,使用的核磁仪器为brukerav250。所用的硅胶柱为200-300目与40-63目。反应机理为阴离子反应,如图2所示。本步骤产率为35.4%,反应产物熔点为156.2℃。
如图11所示,本发明2,6-二溴-9,10-双[2-[三(1-甲基乙基)甲硅烷基乙炔]蒽具有较高的荧光特性,其紫外-可见光谱图证明其荧光特性,使用的仪器为hitachiu-2010beamuv/visible,其熔点测试表明其具有156.2℃的高熔点,表明其具有较高的稳定性,适合于较大温度差的环境下的应用,具有较好的抗干扰能力。
图8为2,6-二溴-9,10-双[2-[三(1-甲基乙基)甲硅烷基乙炔]蒽的质谱图,证实了其分子量696.75。表明其具有较大的分子量,在实际应用中具有较高的价值。
综上所述,本发明使用2,6-氨基蒽醌,通过桑德迈尔(sandmeyer)反应以t-buono为催化剂,将2,6位的氨基取代为溴基。再通过阴离子反应以丁基锂作为引发剂的阴离子反应,将三异丙基硅基乙炔加成在2,6-溴基蒽醌的9,10位上,并使用氯化亚锡将9,10位形成的羟基切除,通过硅胶色谱柱200-300目与40-63目各进行提纯一次将所得产物收集,得到2,6-二溴-9,10-双[2-[三(1-甲基乙基)甲硅烷基乙炔]蒽;过程简单,用时较短,具有很好的重现性,实际运用价值高;本发明的荧光化合物较好荧光效果的分子结构设计符合荧光物质应用需求。本发明的生产制作方法较为简单,成本低廉,操作工艺简单;本发明的荧光化合物,具有荧光效果好,熔点高,稳定性好,可长期室温保存的特点;合成工艺路线简单清晰,可重现性高,生产过程节约时间与试剂,具有较高的工业生产与实际应用价值。
上面对本专利的较佳实施方式作了详细说明,但是本专利并不限于上述实施方式,在本领域的普通技术人员所具备的知识范围内,还可以在不脱离本专利宗旨的前提下做出各种变化。
- 还没有人留言评论。精彩留言会获得点赞!
- 化合物及使用其的有机电激发光组件的制作方法与工艺
- 一种烟草中倍半萜类化合物的测定方法与流程
- 对有机含磷化合物有毒气体具有荧光响应的一维有机半导体纳米线/带及其制备方法和应用与流程
- 一种用于调节白光LED的荧光化合物及其制备方法和应用与流程
- 联咔唑化合物、光固化性组合物、其固化物、用于塑料透镜的固化性组合物和塑料透镜的制作方法与工艺
- 联咔唑化合物、光固化性组合物、其固化物、用于塑料透镜的固化性组合物和塑料透镜的制作方法与工艺
- 一种基于π‑π堆积化合物的荧光材料及其制备方法与流程
- 一种有机电致荧光化合物及其在电致发光器件中的应用的制作方法与工艺
- 一种有机电致荧光化合物及其在电致发光器件中的应用的制作方法与工艺
- 一种有机荧光化合物及其应用的制作方法与工艺