一种利用Cas9/gRNA系统构建扩增子文库的方法与流程
本发明属于高通量测序领域,具体而言,本发明涉及一种构建扩增子文库的方法。
背景技术:
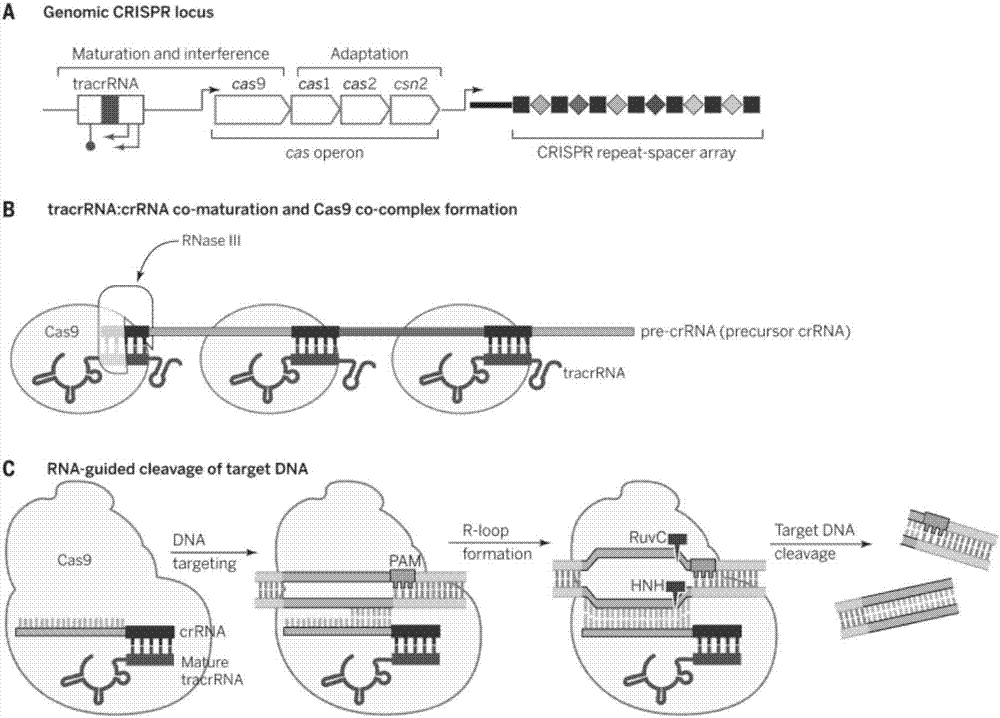
:多重pcr靶向捕获测序技术是指,利用多重pcr技术同时对基因组dna上的多个目标区域进行扩增,得到扩增子,然后通过酶连接或者通过pcr方式将二代测序接头添加到扩增子序列的两侧,得到扩增子文库,之后进行二代测序,获取目标区域的序列信息。多重pcr靶向捕获技术与液相杂交捕获测序技术相比,具有以下优势:1.灵敏度高、特异性强、文库构建所需样本量低;2.文库构建周期短,一次文库构建的通量高,文库构建的成本低;3.测序深度高,适合对热点区域进行测序分析。基于以上优势,多重pcr靶向捕获测序技术已受到诸多测序公司的重视,基于该技术开发出的测序产品在测序市场上广受欢迎和好评,并且需求量逐年上升。因此,多重pcr靶向捕获技术已经成为靶向测序技术中的重要一员,在推进全民享受低成本,高质量的靶向测序服务的道路上,发挥着越来越重要的作用。尽管多重pcr靶向捕获测序技术的优势明显,但是该技术仍然存在一些不足,这严重制约了该技术的应用。在利用多重pcr技术进行靶向捕获测序时,获取的数据通常捕获率低、覆盖率低、均一性差,难以满足客户的要求。究其原因,主要是多重pcr反应过程中会产生大量的引物二聚体。引物二聚体不但会直接与目标区域的扩增子竞争引物和dna聚合酶,同时还会加速底物的消耗,从而严重抑制目标区域的扩增。此外,引物二聚体由于片段小,比扩增子更有利于扩增,因此在测序时,引物二聚体更易发生桥式pcr反应成簇,导致测序数据中含有大量无效数据,增加测序的成本。这些问题随着多重捕获区域数目的增多而加剧,已经成为多重pcr靶向捕获测序应用道路上的拦路虎。因此,本领域需要改进的多重pcr技术用于靶向捕获测序。技术实现要素:为了克服现有技术中存在的问题,本发明提供了一种构建扩增子文库的方法,该方法能够有效去除文库构建过程中产生的引物二聚体,提高测序数据的捕获率,覆盖率和均一性。因此,在第一方面中,本发明提供了一种构建扩增子文库方法,包括以下步骤:s1:采用多重pcr引物对多个目标区域进行扩增,得到多个扩增子;s2:采用cas9/grna系统对所述扩增子进行消化,特异性识别和切割扩增子和引物二聚体两侧的通用序列,去除引物二聚体;s3:依次对所述扩增子进行5’端磷酸化修饰,3’端添加“a”,然后用连接酶将测序接头例如p5和p7连接到扩增子的5’端和3’端;s4:将所述连接后的扩增子磁珠纯化后,得到扩增子文库,然后进行二代测序。在优选的实施方案中,在所述方法的步骤s1中,所述多重pcr引物的3’端为特异性序列,能够与模板互补配对;所述多重pcr引物的5’端为通用序列。在优选的实施方案中,在所述方法的步骤s1中,采用多重pcr引物对多个目标区域进行扩增,所述多重pcr引物的3’端是与目标区域互补配对的特异性序列,5’端是一段长度在23nt以上的通用序列,其3’端的最后三个碱基通常为ngg。ngg序列前面的20碱基序列与cas9/grna系统中grna5’端的20碱基序列相同。在优选的实施方案中,在所述方法的步骤s1和步骤s2之间,采用磁珠对多重pcr扩增产物进行纯化,去除引物,dna聚合酶,dntp等物质,得到高纯度的扩增子和引物二聚体。在优选的实施方案中,在所述方法的步骤s2中,采用cas9/grna系统对扩增子和引物二聚体两侧的通用序列进行特异性切割,去除引物二聚体和扩增子两侧的通用序列,去除引物二聚体。优选地,在所述方法的步骤s2中,采用cas9蛋白酶和grna,对扩增子和引物二聚体进行消化处理,特异性切割掉扩增子和引物二聚体两侧的通用序列,去除引物二聚体。所述cas9蛋白酶包含多种cas9蛋白酶的变体,如切割双链、切割单链以及识别不同pam结构。在优选的实施方案中,在步骤s2和s3中,采用磁珠对s2步骤的酶切产物进行纯化,得到扩增子。在优选的实施方案中,grna的5’端20碱基序列必须与多重pcr引物的5’端通用序列中的ngg序列前面的20个碱基序列一致。grna的来源包括化学合成和体外转录。如果体外使用t7dna聚合酶转录grna,grna的5’端要求添加gg序列,从而被t7dna聚合酶识别。如果使用t4dna聚合酶进行转录,在grna的5’端通常添加x碱基,增加t4dna聚合酶的识别和转录。grna加入到反应体系中的种类与多重pcr引物使用的通用序列种类一致。在优选的实施方案中,在步骤s3中,对扩增子依次进行5’端磷酸化修饰,3’端添加“a”,然后用连接酶将测序接头例如p5和p7连接到扩增子的5’端和3’端。每一步操作结束后,均采用磁珠纯化扩增子。在优选的实施方案中,所述操作步骤s3中,对所述扩增子的5’端进行磷酸化处理,对扩增子的3’端添加“a”,并采用连接酶将测序接头(例如p5和p7)分别连接到所述扩增子的5’端,磁珠纯化后,得到扩增子文库,进行二代测序。在优选的实施方案中,在所述方法的步骤s1、s2和s3后,以及步骤s3中的扩增子5’端磷酸化修饰、扩增子3’端添加“a”、扩增子5’端和3’端分别连接接头序列例如p5和p7等操作后,均采用磁珠对所述扩增子进行纯化。在第二方面中,本发明提供了一种利用cas9/grna系统构建的扩增子文库,所述扩增子文库使用本发明第一方面的方法构建而成。附图说明通过以下附图对本发明进行说明图1:基于cas9/grna系统构建扩增子文库的流程图;图2:crispr/cas9的元件及工作模式图[1]。图3cas9/grna系统构建300重扩增子文库的质检结果。具体实施方式本发明提供了一种构建扩增子文库构建的方法,该方法能够有效去除文库构建过程中产生的引物二聚体,提高测序数据的捕获率,覆盖率和均一性。在本发明中,在所述方法的步骤s1中,所述多重pcr引物的3’端为特异性序列,能够与模板互补配对;所述多重pcr引物的5’端为通用序列。在本发明中,所述多重pcr引物5’端通用序列的长度在23nt以上,且通用序列3’端存在ngg的碱基排列,ngg序列前面的20个碱基序列与cas9/grna系统中grna5’端的20个碱基序列一致。在本发明中,使用多种不同的通用序列,在s2步骤中加入与之序列一致的grna。在本发明中,所述方法的步骤s2中,采用cas9蛋白酶和grna,对扩增子和引物二聚体进行消化处理,特异性切割掉扩增子和引物二聚体两侧的通用序列,去除引物二聚体。在本发明中,所述cas9蛋白酶包含多种cas9蛋白酶的变体,如所述变体切割双链、切割单链以及识别不同pam结构。在本发明中,grna的5’端的20个碱基序列与所述多重pcr引物5’端通用序列中的ngg序列前面的20个碱基序列一致。在本发明中,grna的来源包括化学合成和体外转录,如果体外使用t7dna聚合酶转录grna,grna的5’端要求在原有序列的基础上的添加gg序列,从而被t7dna聚合酶识别;如果使用t4dna聚合酶进行转录,在grna的5’端要求在原有序列的基础上添加x碱基,增加t4dna聚合酶的识别和转录。在本发明中,grna加入到反应体系中的种类与多重pcr引物使用的通用序列种类一致。在本发明中,在所述方法的步骤s3中,对所述扩增子的5’端进行磷酸化处理,对扩增子的3’端添加“a”,并采用连接酶将测序接头列入p5和p7分别连接到所述扩增子的5’端,磁珠纯化后,得到扩增子文库,进行二代测序。在本发明中,在所述方法的步骤s1、s2和s3后,以及步骤s3中的扩增子5’端磷酸化修饰、扩增子3’端添加“a”、扩增子5’端和3’端分别连接接头序列(例如p5和p7)等操作后,均采用磁珠对所述扩增子进行纯化。虽然不希望拘囿于任何理论,但发明人认为crispr/cas9(clusteredregularlyinterspacedpalindromicrepeat)系统可能对本发明的有益效果有促进作用。crispr/cas9(clusteredregularlyinterspacedpalindromicrepeat)系统[1]是发现于细菌中的一种选择性免疫系统,能够特异性识别并切割侵入细菌内的外源gdna,从而保护细菌。crispr/cas9系统主要由三部分组成:tracrrna、cas9蛋白和crrna。tracrrna的核酸序列位于cas9基因的上游,转录后经过iii型核酸酶的作用而成熟,其主要功能与crrna中的重复序列互补配对,介导crrna成熟。cas9蛋白具有两个重要的结构域,分别为hnh和ruvc。hnh切割crrna与靶dna杂交形成的双链,ruvc切割靶dna解开后未配对的单链。此外,nhn和ruvc由一段富含精氨酸的α螺旋链接,该铰链区主要负责与grna-targetdna杂合体结合。crrna的核酸序列由一连串的短重复序列被不同特异序列分隔形成。短重复序列主要与tracrrna结合,特异序列来源于质粒或者外源gdna,能够与外源靶dna互补配对,从而对靶dna进行特异性识别和切割。crispr/cas9识别的靶序列要求在其3’端存在pam序列,其碱基序列通常为ngg。pam序列对于crispr/cas9识别靶dna非常重要,缺失pam结构将导致crispr/cas9无法识别和切割靶dna序列。crispr/cas9的工作流程主要包括以下几个:1.crispr/cas9复合体识别靶dna上游的pam结构,并引发靶dna解链,形成r-loop结构;2.crrna5’的特异性序列(20nt)与解链的靶dna互补配对,形成rna-dna二聚体结构;3.cas9蛋白中hnh结构域在pam上游的第3个碱基处切割rna-dna二聚体,ruvc结构域也在pam序列上游的第3个碱基处切割靶dna中未配对的单链,从而将双链靶dna切断,形成平末端;4.切断的双链通过非同源末端链接(nhej)或者同源修复,便可在切割的位置引入突变,导致靶dna的序列发生改变。研究人员通过分子生物学技术,将tracrrna和crrna融合成一条grna(guiderna),进一步简化了crispr/cas9编辑靶基因的操作流程。目前,crispr/cas9因其操作简便,特异性高,已经成为靶基因编辑的主流工具。本发明充分利用cas9/grna系统特异性识别和切割靶dna序列的特性,在多重pcr引物的5’端添加一段cas9/grna系统识别的通用序列,经第一轮多重pcr反应后,采用cas9/grna系统对扩增子和引物二聚体两侧的通用序列进行识别和切割,去除引物二聚体,随后对扩增子进行磁珠纯化,5’端进行磷酸化修饰,3’端添加“a”,采用连接酶将测序接头(例如p5和p7)分别连接到扩增子的5’端和3’端,磁珠纯化后得到扩增子文库,进行上机测序。实施例1利用该方法构建单个反应管300重扩增子文库的实施例1如下所示。第1步:300重pcr反应扩增靶向产物1.300重pcr的反应体系如下表1所示:dna聚合酶,缓冲液和dntp均为neb的产品(货号m0493l);模板是从293t细胞提取得到的gdna;多重引物的浓度为5mm,引物序列分为5’端的通用序列和3’端的特异性序列。所有上游引物5’端的通用序列为5’tctgtctatgacgctgtatccgg3’,所有下游引物5’端的通用序列为5’atccactgacatagtctgtatgg3’。300对多重引物3’端的特异性序列如表2所示。表1300重pcr的反应体系试剂体积(μl)无核酸酶水9.55×q5反应缓冲物510mmdntps0.5引物混合物(5mm)4模板dna(10ng/μl)5q5hotstarthigh-fidelitydna聚合酶(2u/μl)1表2300重引物3’端的特异性序列(seqidno.1-600)1.2300重pcr反应按照下表3进行操作表3300重pcr的反应条件第2步:对第1轮的300重pcr产物进行磁珠纯化2.1向25μl300重pcr产物中加入40μl室温平衡后的ampurexp磁珠,用移液器吸打混匀数次;2.2室温孵育10min后,将pcr管置于dynamag-96side磁力架上3min;2.3移除上清,pcr管继续放置在磁力架上,向管内加入180μl80%乙醇溶液,静置30s;2.4移除上清,再加入180μl80%乙醇溶液,静置30s后彻底移除上清(使用10μl移液器移除底部残留乙醇溶液);2.5室温静置10min,使残留乙醇彻底挥发;2.6将pcr管从磁力架取下,加入22μl无核酸酶水,移液器轻轻吸打重悬磁珠,避免产生气泡,室温静置2min;2.7将pcr管重新置于磁力架上,静置4min;2.8用移液器吸取20μl上清液,转移到新的200μlpcr管内,管内上清液为多重pcr扩增产物。第3步:grna的体外转录本实施例采用neb公司的sgrna体外转录试剂盒(engentmsgrnasynthesiskit)对靶rna进行体外转录。由于在多重pcr反应中,上下游引物5’端的通用序列不同,因此需要体外转录2条grna。第1条grna(编号为grna1)的体外转录模板为5’ttctaatacgactcactatagtctgtctatgacgctgtatc3’,得到的grna1与cas9蛋白(本实施例采用的cas9蛋白为neb公司的产品(货号m0646))结合后,可以识别并切割扩增产物和引物二聚体中上游引物5’端的通用序列;第2条grna(编号为grna2)的体外转录模板为5’ttctaatacgactcactatagatccactgacatagtctgta3’,得到的grna2与cas9蛋白结合后,可以识别并切割扩增产物和引物二聚体中下游引物5’端的通用序列。3.1grna1和grna2的体外转录反应体系如下表4所示:表4grna体外转录合成的反应体系试剂体积(μl)无核酸酶水3engen2xsgrnareactionmix,s.pyogenes10target-specifcdnaoligo(1μm)5engensgrnaenzymemix2备注:target-specifcdnaoligo为grna1和grna2的体外转录模板3.2将上述反应试剂加入到200μl的pcr反应管之后,37℃孵育30min,然后置于冰盒上暂存;3.3向上述反应体系内加入30μl无核酸酶水,然后加入2μldnasei,振荡混匀离心后,37℃孵育15min,去除反应体系内的dna模板。第4步:grna的纯化第3步体外转录合成的grna使用zymoresearch公司的rna纯化试剂盒(rnaclean&concentratortm-25)进行纯化,具体操作步骤如下:4.1加入100μlrnabindingbuffer到50μlgrna体外转录产物中,振荡混匀;4.2加入150μl100%的乙醇到上述反应体系中,振荡混匀;4.3将离心柱安放于收集管上,并将上述反应液转移到离心柱中(zymo-spintmiiccolumn),12000g,离心30s,舍弃收集液;4.4加入400μlrnaprepbuffer到离心柱中,离心30s,舍弃收集液;4.5加入700μlrnawashbuffer到离心柱中,离心30s,舍弃收集液;4.6加入400μlrnawashbuffer到离心柱中,离心2min,将离心柱转移到rnase-free的1.5ml离心管中;4.7加入25μlrnase-free的ddh2o于离心柱的核酸吸附膜上,离心30s,得到纯化后的grna。第5步:cas9/grna消化多扩增子和引物二聚体两侧5’端的通用序列,去除引物二聚体5.1本实施例采用的cas9蛋白为neb公司的产品(货号m0646),首先将cas9蛋白与grna1和grna2混合孵育,制备cas9/grna1和cas9/grna2复合体。反应体系如下表5所示。表5cas9/grna1和cas9/grna2复合体制备的反应体系试剂体积(μl)无核酸酶水1310×cas9nucleasereactionbuffer5grna1(300nm)5grna2(300nm)5cas9nuclease,s.pyogenes(1μm)25.2将上述反应试剂混合后,25℃孵育10min;5.3向上述反应体系内加入20μl第2步纯化得到的多重pcr产物,总体积为50μl,然后混匀离心,37℃孵育2h,消化多重pcr产物及引物二聚体两侧5’端的通用引物序列,去除引物二聚体;第6步:磁珠纯化第5步的消化产物6.1向50μl消化产物中加入75μl室温平衡后的ampurexp磁珠,用移液器吸打混匀数次;6.2室温孵育10min后,将pcr管置于dynamag-96side磁力架上3min;6.3移除上清,pcr管继续放置在磁力架上,向管内加入180μl80%乙醇溶液,静置30s;6.4移除上清,再加入180μl80%乙醇溶液,静置30s后彻底移除上清(使用10μl移液器移除底部残留乙醇溶液);6.5室温静置10min,使残留乙醇彻底挥发;6.6将pcr管从磁力架取下,加入32μl无核酸酶水,移液器轻轻吸打重悬磁珠,避免产生气泡,室温静置2min;6.7将pcr管重新置于磁力架上,静置4min;6.8用移液器吸取30μl上清液,转移到新的200μlpcr管内,管内上清液为去除两侧通用引物序列的多重pcr产物;第7步:对扩增子的5’端添加磷酸基团7.1本实施例中,对多重pcr产物5’端添加磷酸基团的试剂为kapabiosystems公司的产品(货号kk8234),反应体系如下表6所示:表6扩增子5’端磷酸化修饰的反应体系试剂体积(μl)水3endrepairbuffer(10x)4endrepairenzymemix3多重pcr产物30备注:多重pcr产物为第6步纯化得到的多重pcr产物7.2将上述反应试剂混合后,振荡混匀,离心,20℃孵育30min;第8步:对5’端添加磷酸基团的扩增子进行磁珠纯化8.1向40μl5’端添加磷酸基团后的多重pcr产物加入60μl室温平衡后的ampurexp磁珠,用移液器吸打混匀数次;8.2室温孵育10min后,将pcr管置于dynamag-96side磁力架上3min;8.3移除上清,pcr管继续放置在磁力架上,向管内加入180μl80%乙醇溶液,静置30s;8.4移除上清,再加入180μl80%乙醇溶液,静置30s后彻底移除上清(使用10μl移液器移除底部残留乙醇溶液);8.5室温静置10min,使残留乙醇彻底挥发,得到的磁珠上有5’端磷酸化后的多重pcr产物,直接作为下一步反应的底物;第9步:对扩增子的3’端添加“a”9.1本实施例采用的试剂为kapabiosystems公司的产品(货号kk8234),反应体系如下表7所示:表7扩增子3’端添加“a”的反应体系9.2将上述反应试剂混合后,振荡混匀,30℃孵育30min;第10步:对3’端添加“a”后扩增子进行磁珠纯化10.1向上述50μl反应体系内,加入90μlpeg/nacl溶液,用移液器上下吹打混匀;10.2室温孵育10min后,将pcr管置于dynamag-96side磁力架上3min;移除上清,pcr管继续放置在磁力架上,向管内加入180μl80%乙醇溶液,静置30s;10.3移除上清,再加入180μl80%乙醇溶液,静置30s后彻底移除上清(使用10μl移液器移除底部残留乙醇溶液);10.4室温静置10min,使残留乙醇彻底挥发,磁珠结合的扩增子,直接作为下一步反应的底物;第11步:将5’端含有磷酸基团和3’端添加“a”后的扩增子与illumina平台的测序接头连接,得到扩增子文库11.1本实施例中采购的dna连接酶为kapabiosystems的产物(货号kk8235)。连接的反应体系如下表8所示。表8扩增子与测序接头序列连接的反应体系11.2将上述反应试剂混合后,振荡混匀,20℃孵育15min;第12步:对连接后的扩增子文库进行磁珠纯化12.1向上述50μl反应体系内,加入50μlpeg/nacl溶液,用移液器上下吹打混匀;12.2室温孵育10min后,将pcr管置于dynamag-96side磁力架上3min;12.3移除上清,pcr管继续放置在磁力架上,向管内加入180μl80%乙醇溶液,静置30s;12.4移除上清,再加入180μl80%乙醇溶液,静置30s后彻底移除上清(使用10μl移液器移除底部残留乙醇溶液);12.5室温静置10min,使残留乙醇彻底挥发;12.6将pcr管从磁力架取下,加入22μl无核酸酶水,移液器轻轻吸打重悬磁珠,避免产生气泡,室温静置2min;12.7将pcr管重新置于磁力架上,静置4min;12.8用移液器吸取20μl上清液,转移到新的200μlpcr管内,管内上清液为多重pcr扩增产物。第13步:对纯化后的扩增子文库进行pcr扩增,提高扩增子文库的浓度13.1本实施例采用的引物和dna聚合酶均为kapabiosystems公司的产品(货号kk2600)。pcr的反应体系如下表9所示:表9扩增子文库扩增的反应体系试剂体积(μl)kapahifihotstartreadymix(2x)25libraryamplifcationprimermix(10x)5adapter-ligatedlibrarydna2013.2将上述反应成分混合后,震荡混匀,离心,然后按照下表10进行pcr反应扩增;表10扩增子文库pcr的反应条件第14步:对pcr产物进行磁珠纯化,得到高浓度的文库14.1向50μlpcr产物中加入70μl室温平衡后的ampurexp磁珠,用移液器吸打混匀数次;14.2室温孵育10min后,将pcr管置于dynamag-96side磁力架上3min;14.3移除上清,pcr管继续放置在磁力架上,向管内加入180μl80%乙醇溶液,静置30s;14.4移除上清,再加入180μl80%乙醇溶液,静置30s后彻底移除上清(使用10μl移液器移除底部残留乙醇溶液);14.5室温静置10min,使残留乙醇彻底挥发;14.6将pcr管从磁力架取下,加入22μl无核酸酶水,移液器轻轻吸打重悬磁珠,避免产生气泡,室温静置2min;14.7将pcr管重新置于磁力架上,静置4min;14.8用移液器吸取20μl上清液,转移到新的200μlpcr管内,管内上清液为多重pcr扩增产物。第15步:对扩增子文库进行浓度测量取2μl文库使用3.0fluorometer(qubitdsdnahsassaykit)进行浓度测量,得到的文库浓度为7.84ng/μl;第16步:对扩增子文库的片段分布进行质检取2μl扩增子文库使用qsep100全自动核酸蛋白分析系统(厚泽生物)进行质检,得到的质检峰图如图3所示:文库的靶条带位置正确,分布250-420bp之间,主峰位置在356bp;文库中基本没有引物二聚体。第17步:对文库进行二代测序并进行数据分析采用illumina公司的next-seq500测序平台对上述文库进行二代测序,对得到的测序数据进行统计分析,结果如下表11所示:文库的碱基质量评分(%)为90.12%,比对率为99.56%,覆盖率为100%,20%平均测序深度为98.84%,均一性高。文库的各项数据指标表现优异。表11cas9/grna系统构建300重扩增子文库的测序数据统计结果参考文献:[1]doudnaja,charpentiere.genomeediting.thenewfrontierofgenomeengineeringwithcrispr-cas9.science.2014nov28;346(6213):1258096.。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1