一种β‑咔啉化合物及其合成方法和应用与流程
【
技术领域:
】本发明涉及医药
技术领域:
,具体涉及一种β-咔啉化合物及其合成方法和应用。
背景技术:
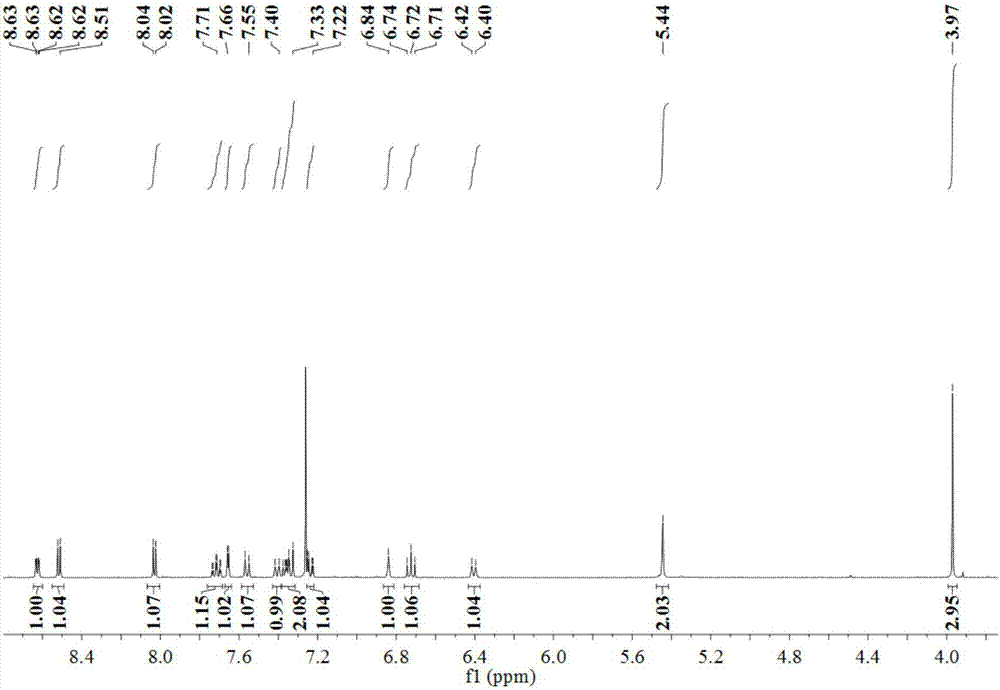
:慢性肾脏病以肾功能进行性丧失、细胞外基质(ecm)大量聚积所导致的肾纤维化为特征,主要包括肾小球硬化和小管间质纤维化。肾间质纤维化几乎是所有慢性肾脏疾病发展的最终结果,与肾功能衰竭进展密切相关,是导致终末期肾衰竭的主要病因。若早期对肾脏纤维化进行干预治疗,将有助于延缓慢性肾脏病进展,极大减少终末期肾病的发生率及其并发症,减轻其所造成的社会经济负担。研究发现,转化生长因子-β(tgf-β)及其介导的smad信号通路在肾间质纤维化中起重要作用,被认为是最关键、最强的纤维化诱导因子,参与肾纤维化的各个环节中。它的过度表达能够刺激系膜细胞、间质成纤维细胞合成大量胶原蛋白(collagen)、纤连蛋白(fn)和层粘连蛋白(ln),刺激成纤维细胞增殖并活化成肌成纤维细胞,促进ecm的分泌与沉积。tgf-β还能介导肾小管上皮细胞向间质细胞的转分化(emt)。目前临床上尚无有效的抗肾脏纤维化的治疗方法。拮抗tgf-β及其介导的信号通路,抑制ecm合成或促进其降解,可能为寻找治疗肾脏纤维化提供一个有效的途径。β-咔啉类生物碱是从新疆药用植物骆驼蓬(peganumharmala)中分离出来的一种活性成分。具有广谱的药理学活性,如抗焦虑、抗抑郁、抗痉挛、以及抗肿瘤、抗疟疾、抗寄生虫、抗艾滋病等。在中草药配方中,用骆驼蓬种子来治疗癌症由来已久。β-咔啉类生物碱的结构修饰一直备受科研工作者的关注。一般认为,大多数β-咔啉类生物碱的平面共轭结构以及不同取代基团(如苄基(-c6h5)、甲氧基(-och3))的存在与其抗肿瘤活性具有显著关系。目前对β-咔啉类生物碱的研究主要包括以下几个方面:一是基于天然产物化学研究从不同科属的植物中发现并进行分离提纯;二是通过有机合成方法对β-咔啉类生物碱进行全合成或半合成,对其结构进行修饰。另外则是对其药理活性(如抗肿瘤活性)进行分子机理研究。如插入dna,抑制拓扑异构酶、抑制cdk等。但从目前的研究进展来看,β-咔啉类生物碱在天然植物中的含量一般都比较低,而且提取相对比较复杂,不利于对其进行深入研究,而有机半合成方法在产率上虽具有一定的优势,但受到制取成本的限制,因此目前对β-咔啉类生物碱的拓展研究尚显不足。该类化合物有着良好的发展前景,但是目前还未见有1-吡啶-6-甲氧基-9-(4-甲基苄)β-咔啉及其合成和应用的相关报道。技术实现要素:本发明的发明目的在于:针对上述存在的问题,提供一种β-咔啉化合物及其合成方法和应用。本发明生产过程环保,生产成本低,操作安全性高,反应条件温和,操作简单;得到的β-咔啉化合物可用于制备抗肾纤维化药物和/或抗慢性肾病药物。为了实现上述目的,本发明采用的技术方案如下:一种式i的化合物或其药学上可接受的盐:上述式i所示化合物的化学名称为:1-吡啶-6-甲氧基-9-(4-甲基苄)β-咔啉。分子量为:379.17。本发明还提供了所述式i的化合物的合成方法,包括如下步骤:将氢化钠溶于n,n-二甲基甲酰胺中,然后加入式ⅱ所示化合物1-吡啶-6-甲氧基-β-咔啉和4-甲基苄基溴,于常温或加热条件下一锅反应,即得所述式i的化合物。上述合成方法的合成路线如下:上述式i所示化合物合成方法的步骤中,原料1-吡啶-6-甲氧基-β-咔啉和另一原料4-甲基苄基溴的物质的量之比通常为1:1.5或1:1,优选4-甲基苄基溴的摩尔量稍大于1-吡啶-6-甲氧基-β-咔啉的物质的量,以使反应充分进行。进一步地,所述反应在40℃至有机溶剂的回流温度下进行。反应是否完全可以采用薄层层析(tlc)跟踪检测,通常控制反应时间为20min~2h较合适。反应的方式优选采用回流反应,当采用回流反应时,反应更优选在有机溶剂的回流温度下进行。本发明所述的合成方法,还包括分离和纯化步骤,所述分离和纯化为将一锅反应所得产物滴加碱性溶液调至中性或碱性,用乙酸乙酯或氯仿萃取,收集有机相,除去溶剂,然后采用硅胶柱层析或重结晶的方式来进行纯化;当采用硅胶柱层析的方式进行纯化时,以体积比为1000:50~500的石油醚和乙酸乙酯组成的洗脱剂进行洗脱,洗脱液除去溶剂,得到纯化后的目标物。本发明还提供了所述的化合物或其药学上可接受的盐在制备抗肾纤维化药物和/或抗慢性肾病药物中的应用。本发明还提供了一种药物组合物,其包括治疗有效量的权利要求1所述的化合物或其药学上可接受的盐以及药学上可接受的辅料。综上所述,由于采用了上述技术方案,本发明的有益效果是:与现有技术相比,本发明提供了一种β-咔啉化合物及其合成方法和应用。发明人对本发明所述化合物的抗肾纤维化、抗慢性肾病活性研究表明,该化合物体外抗肾纤维化活性显著,具有较好的潜在药用价值,可用于各种抗肾纤维化、抗慢性肾病药物的制备。【附图说明】图1为本发明实施例1制得的最终产物的核磁共振氢谱图;图2为本发明实施例1制得的最终产物的核磁共振碳谱图;图3为本发明实施例1制得的最终产物的电喷雾质谱谱图;图4为本发明实施例1制得的最终产物的细胞蛋白质免疫印迹实验图。【具体实施方式】实施例1结构如式i所示化合物的制备1)将200mg(8.3mmol)的nah,溶于1mln,n-二甲基甲酰胺中,搅拌10min,并逐渐加入275mg(1mmol)的1-吡啶-6-甲氧基-β-咔啉,室温搅拌10min,然后再逐滴加入277.5mg(1.5mmol)的4-甲基苄基溴,常温继续反应约20min;反应结束后所得产物用300ml的氨水调节至中性,并用乙酸乙酯萃取,收集有机相,减压除去溶剂,得黄色粉末的粗产物;2)粗产物以石油醚:乙酸乙酯=1000:100(体积比)组成的溶剂作为洗脱剂,硅胶柱层析,得到淡黄色粉末产物(245mg,产率64.6%,纯度99.92%)。对所得淡黄色粉末产物进行核磁表征,其核磁共振氢谱和碳谱分别如图1和2所示。1hnmr(500mhz,cdcl3)δ8.70–8.65(m,1h),8.55(d,j=5.2hz,1h),8.08(d,j=5.2hz,1h),7.71(dd,j=7.6,1.7hz,1h),7.66(t,j=5.1hz,2h),7.38(d,j=9.0hz,1h),7.36–7.32(m,1h),7.25(dd,j=9.0,2.5hz,1h),6.84(d,j=7.9hz,2h),6.41(d,j=8.0hz,2h),5.46(s,2h),3.98(s,3h),2.22(s,3h).13cnmr(126mhz,cdcl3)δ154.44,148.19,138.39,136.76,136.70,134.60,134.09,128.97,125.52,125.36,123.26,121.42,119.28,114.67,111.60,103.22,77.29,77.24,77.04,76.78,56.01,48.58,20.99.对所得淡黄色粉末产物进行质谱分析,结果分别如图3所示。因此,可确定上述淡黄色粉末产物为:1-吡啶-6-甲氧基-9-(4-甲基苄)β-咔啉,其化学结构式如下式i所示:实施例2结构如式i所示化合物的制备将400mg(16.6mmol)的nah,溶于1mln,n-二甲基甲酰胺中,搅拌10min,并逐渐加入550mg(2mmol)的1-吡啶-6-甲氧基-β-咔啉,室温搅拌10min,然后再逐滴加入555mg(3mmol)的4-甲基苄基溴,常温继续反应约20min;反应结束后所得产物用用400ml氨水调节至中性,并用乙酸乙酯萃取,收集有机相,减压除去溶剂,得黄色油状液体的粗产物;粗产物以石油醚:乙酸乙酯=1000:200(体积比)组成的溶剂作为洗脱剂,硅胶柱层析,得到淡黄色粉末产物(470mg,产率62.0%,纯度99.91%)。所得淡黄色粉末产物经过核磁表征,确定为本发明所述的1-吡啶-6-甲氧基-9-(4-甲基苄)β-咔啉。实施例3结构如式i所示化合物的制备1)将200mg(8.33mmol)的nah,溶于5mln,n-二甲基甲酰胺中,搅拌10min,并逐渐加入275mg(1mmol)的1-吡啶-6-甲氧基-β-咔啉,室温搅拌10min,然后再逐滴加入370mg(2mmol)的4-甲基苄基溴,常温继续反应约20min;反应结束后所得产物用用400ml氨水调节至中性,并用乙酸乙酯萃取,收集有机相,减压除去溶剂,得黄色油状液体的粗产物;2)粗产物以石油醚:乙酸乙酯=1000:50(体积比)组成的溶剂作为洗脱剂,硅胶柱层析,得到淡黄色粉末产物(134mg,产率35.4%,纯度99.93%)。所得淡黄色粉末产物经过核磁表征,确定为本发明所述的1-吡啶-6-甲氧基-9-(4-甲基苄)β-咔啉。实施例4结构如式i所示化合物的制备1)将200mg(8.33mmol)的nah,溶于8mln,n-二甲基甲酰胺中,搅拌10min,并逐渐加入275mg(1mmol)的1-吡啶-6-甲氧基-β-咔啉,室温搅拌10min,然后再逐滴加入185mg(1mmol)的4-甲基苄基溴,常温继续反应约20min;反应结束后所得产物用用400ml氨水调节至中性,并用乙酸乙酯萃取,收集有机相,减压除去溶剂,得黄色油状液体的粗产物;2)粗产物以石油醚:乙酸乙酯=1000:300(体积比)组成的溶剂作为洗脱剂,硅胶柱层析,得到淡黄色粉末产物(211mg,产率55.7%,纯度99.90%)。所得淡黄色粉末产物经过核磁表征,确定为本发明所述的1-吡啶-6-甲氧基-9-(4-甲基苄)β-咔啉。实施例5结构如式i所示化合物的制备1)将200mg(8.33mmol)的nah,溶于10mln,n-二甲基甲酰胺中,搅拌10min,并逐渐加入275mg(1mmol)的1-吡啶-6-甲氧基-β-咔啉,室温搅拌20min,然后再逐滴加入370mg(2mmol)的4-甲基苄基溴,常温继续反应约40min;反应结束后所得产物用用500ml氨水调节至中性,并用乙酸乙酯萃取,收集有机相,减压除去溶剂,得黄色油状液体的粗产物;2)粗产物以石油醚:乙酸乙酯=1000:500(体积比)组成的溶剂作为洗脱剂,硅胶柱层析,得到淡黄色粉末产物(127mg,产率33.5%)。所得淡黄色粉末产物经过核磁表征,确定为本发明所述的1-吡啶-6-甲氧基-9-(4-甲基苄)β-咔啉。实施例6结构如式i所示化合物的制备1)将200mg(8.3mmol)的nah,溶于1mln,n-二甲基甲酰胺中,搅拌10min,并逐渐加入275mg(1mmol)的1-吡啶-6-甲氧基-β-咔啉,室温搅拌10min,然后再逐滴加入277.5mg(1.5mmol)的4-甲基苄基溴,加热至40℃继续反应约20min;反应结束后所得产物用350ml的氨水调节至碱性,并用氯仿萃取,收集有机相,减压除去溶剂,得黄色粉末的粗产物;2)粗产物以石油醚:乙酸乙酯=1000:100(体积比)组成的溶剂作为洗脱剂,硅胶柱层析,得到淡黄色粉末产物(254mg,产率65.2%,纯度99.92%)。所得淡黄色粉末产物经过核磁表征,确定为本发明所述的1-吡啶-6-甲氧基-9-(4-甲基苄)β-咔啉1-吡啶-6-甲氧基-9-(4-甲基苄)β-咔啉。实施例7结构如式i所示化合物1-吡啶-6-甲氧基-9-(4-甲基苄)β-咔啉的抗肾纤维化实验一、1-吡啶-6-甲氧基-9-(4-甲基苄)β-咔啉对大鼠正常肾成纤维细胞nrk-49f的增殖抑制活性实验:1、细胞株与细胞培养本实验选用大鼠正常肾成纤维细胞nrk-49f细胞株。所用细胞株培养在含10wt%小牛血清、100u/ml青霉素、100u/ml链霉素的dmem/f-12培养液内,置37℃含体积浓度5%co2孵箱中培养。2、待测化合物的配制所用的1-吡啶-6-甲氧基-9-(4-甲基苄)β-咔啉为本发明实施例1制得产物,由柱层析提纯所得,纯度≥95%。将待测的受试化合物以dmso溶解后,再用直径d=0.22μm的微孔滤膜过滤除菌,置于4℃下保存。将其dmso储液(浓度为0.002mol/l)通过无血清dmem/f-12培养基依次稀释成五个浓度梯度,分别为20、10、5、2.5、1.25μmol/l,其中助溶剂dmso终浓度≤1%。首先测试不同浓度下的目标产物对于nrk-49f细胞增殖的抑制率,视为初筛结果;再分别测试不同梯度浓度下目标产物对nrk-49f细胞的增殖抑制程度,用以拟合计算半数抑制浓度,即ic50值。3、细胞生长抑制实验(mtt法)(1)取对数生长期的细胞,经胰蛋白酶消化后,用含10%小牛血清的培养液配制成浓度为5000个/ml的细胞悬液,以每孔180μl接种于96孔培养板中,使待测细胞密度至1000~10000个/孔(边缘孔用无菌pbs填充);(2)5%co2,37℃孵育12h,待细胞贴壁后,换无血清dmem/f-12培养液继续培养12h,每孔加入一定浓度梯度的药物20μl,每个浓度梯度设5个复孔;(3)5%co2,37℃孵育48小时,倒置显微镜下观察;(4)每孔加入10μl的mtt溶液(5mg/mlpbs,即0.5%mtt),继续培养4~8h;(5)终止培养,小心吸去孔内培养液,每孔加入200μldmso充分溶解甲瓒沉淀,振荡器混匀后,在酶标仪用波长为570nm,参比波长为450nm测定各孔的光密度值;(6)同时设置调零孔(培养基、mtt、dmso),对照孔(细胞、相同浓度的药物溶解介质、培养液、mtt、dmso)。(7)根据测得的光密度值(od值),来判断活细胞数量,od值越大,细胞活性越强。利用公式:计算化合物对细胞生长的抑制率。其测试结果如以下表1所示。表1:化合物在不同浓度下对nrk-49f细胞株的生长抑制率(%)进一步通过spss软件对三个浓度梯度的抑制率数据进行拟合,求出产物对nrk-49f细胞株的半数抑制浓度(ic50值,单位μmol/l),化合物对于nrk-49f细胞株的ic50值如表2所示。表2:化合物对nrk-49f细胞株的ic50值(μm)从体外测试结果来看,1-吡啶-6-甲氧基-9-(4-甲基苄)β-咔啉对大鼠正常肾成纤维细胞nrk-49f细胞株都表现出显著的增殖抑制活性,用mtt法测得在20、10、5、2.5、1.25μmol/l浓度时抑制率分别为79.88%、57.18%、27.96%、13.11%、0.28%,计算得到ic50值为8.597μm。二、1-吡啶-6-甲氧基-9-(4-甲基苄)β-咔啉对大鼠肾成纤维化细胞nrk-49f的肾纤维化相关蛋白的蛋白质免疫印迹实验:本发明用tgf-β1(2ng/ml)刺激nrk-49f细胞,建立体外纤维化模型,同时加入不同浓度(4μm,8μm,12μm)的上述(i)所示化合物,共同作用48小时后,将细胞裂解后抽提蛋白,进行蛋白质免疫印迹实验,分析纤连蛋白(fibronectin,fn)、α-平滑肌肌动蛋白(α-sma)、胶原蛋白ⅰ(collagenⅰ)等蛋白的表达情况,具体操作如下:1.蛋白提取(1)接种细胞:选择收集生长状态良好的nrk-49f细胞,种于直径为10cm的培养皿,置于37℃,co2浓度为5%的恒温培养箱培养12h,换无血清dmem/f-12培养液继续培养12h。(2)给药:待细胞贴壁增殖达对数生长期时,分别加入2ng/ml的tgf-β1和不同体积、浓度为2×10-3m的化合物ⅰ(由实施例1制得),充分混合均匀后在恒温培养箱中孵育48h。(3)收集细胞:待细胞给药处理48h后,用浓度为0.25%胰蛋白酶消化贴壁细胞,转移至15ml离心管中,转至离心机,2000rpm离心10min。(4)细胞裂解:用细胞裂解液(ripa):pmsf(苯甲基磺酰氟)=100:1的体积比分别加入,混匀悬浮细胞,置于冰上低温环境下裂解30~50min。(5)收集蛋白:将裂解好的细胞悬液低温4℃,12000r离心10min;上清液用bca蛋白浓度测定试剂盒(增强型)进行蛋白定量,-80℃储存。2.sds-page电泳(1)浓缩胶与分离胶的制备:根据实验需要,配制10%、12%分离胶与5%的浓缩胶。将分离胶灌入垂直玻璃槽中,用蒸馏水进行液封。分离胶凝固后,弃去蒸馏水,再将浓缩胶灌入垂直玻璃槽插入梳子,待胶凝固。待分离胶凝固后,拔出梳子,将垂直电泳装置放入电泳槽中等待后续加待测样品进行电泳。(2)上样与电泳:取20μg样品用5×上样缓冲液按体积比为4:1比例稀释,沸水浴中加热5min,冷却后上样,在胶板左侧,加入5μl蛋白预染。加入电泳液,电泳槽灌满,按浓缩胶60v,60min,分离胶80v,160min进行电泳。(3)转膜:取出玻璃板,将凝胶剥离并浸于转膜缓冲液中平衡10min;根据彩色预染蛋白条带进行切胶。取8张滤纸剪裁成与转膜海绵相同大小,在转膜缓冲液中浸湿备用;取合适孔径大小(0.45μm或0.22μm)醋酸纤维素膜(pvdf),剪裁成与切割的page胶相同大小。取8张滤纸剪裁成与转膜海绵相同大小,在转膜缓冲液中浸湿备用;取合适孔径大小(0.45μm或0.22μm)醋酸纤维素膜(pvdf),剪裁成与切割的page胶相同大小。将转膜夹放入转膜槽中,加入转膜缓冲液,恒流200ma,进行转膜电泳。待转膜结束,可用考马斯亮蓝染胶,用丽春红染液染膜,检测转膜效果;pvdf膜用清水洗过后,进行下一步操作。(4)免疫反应:待转膜结束,pvdf膜用pbs漂洗后再用快速封闭液封闭30min。pbst漂洗5min,转至按适当比例配置的一抗纤连蛋白(fibronectin,fn)、α-平滑肌肌动蛋白(α-sma)、胶原蛋白ⅰ(collagenⅰ)和胶原蛋白ⅲ(col3a1))溶液中,4℃孵育过夜。用二抗稀释液稀释二抗,室温下在脱色摇床上孵育约1h,用pbst和pbs漂洗后进行化学发光反应。(5)化学发光:取化学发光试剂(superecl超敏发光液,普利莱基因技术有限公司),与膜蛋白面充分接触,在化学发光成像系统上显影。实验结果如图4所示。从抗肾纤维化测试结果来看,1-吡啶-6-甲氧基-9-(4-甲基苄)β-咔啉可显著抑制tgf-β1诱导的nrk-49f细胞的fn、α-sma、collageni的表达,且呈剂量依赖关系,表明该化合物体外抗肾纤维化活性显著,具有较好的潜在药用价值,可用于各种抗肾纤维化、抗慢性肾脏病药物的制备。实施例10药物组合物本发明提供的式i所示化合物15%淀粉40%微晶纤维素25%羧甲基淀粉纳19%硬脂酸镁1%以上药物和辅料粉碎后过筛,混合即得。实施例11药物组合物本发明提供的式i所示化合物8%淀粉47%微晶纤维素22%羧甲基淀粉纳20%硬脂酸镁3%以上药物和辅料粉碎后过筛,混合即得。实施例11药物组合物本发明提供的式i所示化合物15%淀粉40%微晶纤维素25%羧甲基淀粉纳19%硬脂酸镁1%以上药物和辅料粉碎后过筛,混合后于压片机上压片,即得。实施例11药物组合物本发明提供的式i所示化合物15%干淀粉65%硬脂酸镁20%以上药物和辅料粉碎后过筛,混合后填充入硬明胶胶囊中,即得。实施例12药物组合物本发明提供的式i所示化合物1%氯化钠1%注射用水98%将本发明提供的化合物加氯化钠及适量注射用水,搅拌均匀,加入0.1%针用活性炭,吸附,过滤脱碳,补加注射用水至规定量,微孔过滤膜过滤,按1ml/支灌封,100℃湿热灭菌20min,经灯检合格,即得。实施例13药物组合物本发明提供的式i所示化合物0.1%乙醇0.4%氯化钠0.1%甘露醇0.6%注射用水99%将本发明提供的化合物加乙醇搅拌溶解,加氯化钠、甘露醇及适量注射用水,搅拌均匀,加入0.1%针用活性炭,吸附,过滤脱碳,补加注射用水至规定量,微孔过滤膜过滤,按4ml/支分装,冷冻干燥,封装,经检验合格,即得。上述说明是针对本发明较佳可行实施例的详细说明,但实施例并非用以限定本发明的专利申请范围,凡本发明所提示的技术精神下所完成的同等变化或修饰变更,均应属于本发明所涵盖专利范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1