三维网状埃洛石气凝胶材料及其制备方法与流程
本发明属于纳米材料技术领域,更加具体地说,涉及一种三维网状埃洛石气凝胶材料及其制备方法。
背景技术
埃洛石是天然的粘土矿物中的一种,属高岭石的变种,因此也称为变高岭石。它是由高岭石的片层在自然条件下卷曲而成,在自然界中主要的存在形式是纳米管状(马智,王金叶,高祥,丁彤,秦永宁.埃洛石纳米管的应用研究现状[j].化学进展,2012,(z1):275-283.)。埃洛石矿的分布在全世界各大洲,中国、法国、比利时、新西兰、美国、土耳其等国家都有丰富的储量。埃洛石矿在我国主要分布在广东、湖北、湖南、四川、贵州、云南、山西等省份。
埃洛石是双层1:1型铝硅酸盐材料,具有典型的结晶结构。埃洛石区别于高岭土的本质特征在于埃洛石层间存在或者曾经存在结晶水,埃洛石的片层是由外层的硅氧四面体和内层的铝氧八面体规则排布而成,片层中间是游离的水分子。这些水分子容易脱去,这种脱水过程是不可逆的。埃洛石的外表面主要是si-o-si键组成,内壁则主要是铝羟基(牛继南,强颖怀,王春阳,李祥,周一浩,商翔宇,庄全超.埃洛石的命名、结构、形貌和卷曲机制[j].矿物学报,2014,(01):13-22.)。硅/铝羟基存在于埃洛石的结晶边缘或管的端面上,也有少量的包埋羟基存在于结晶结构的内部。埃洛石含有三种状态的水,主要是吸附水、结晶水和化学水。
埃洛石纳米管具有独特的纳米结构,是一种前景广阔的天然纳米材料。且埃洛石纳米管分布广泛,价格便宜,无毒无害。埃洛石因其独特的纳米结构及管状特性,具有以下优点:首先,来源广泛,价格便宜;埃洛石是一种天然黏土矿物,蕴藏丰富,分布广泛且开采较易。其次,具有很好的生物相容性;埃洛石纳米管自然形成,无毒无害,生物相容性较好。另外,活泼羟基蕴藏在埃洛石表面和层间,利于埃洛石的改性以及进一步应用。再加上本身具有较大的长径比和比表面积、纳米尺度等特点,埃洛石近年来已得到广泛关注和研究。
埃洛石纳米管的应用领域广泛。在陶瓷材料、复合材料、缓释材料、催化剂载体、模板、吸附应用等方面有大量的应用。因为埃洛石纳米管是一种黏土矿,所以可用于陶瓷制作,这属于埃洛石的传统应用领域。埃洛石具有纤维增强功能,是制备超薄精细陶瓷的理想原料。近几年来,对埃洛石/聚合物复合材料及其性能的研究越来越受欢迎。埃洛石能够在多数聚合物复合材料中较好分散,能有效提高聚合物的力学性能、热稳定性、阻燃性以及结晶性能,与其他传统填料相比有较大的优势(伍巍,吴鹏君,何丁,曹贤武,周南桥.埃洛石纳米管在高分子纳米复合材料中的应用进展[j].化工进展,2011,(12):2647-2651+2657.)。埃洛石具有独特结构、环境友好、价廉易得等特点,利用其结构特点和吸附特性能够制备具有新型结构与性能的材料,广泛应用于纳米复合材料领域。
技术实现要素:
本发明克服了现有技术中的不足,提供了一种介孔-微孔埃洛石气凝胶材料及其制备方法,采用具有微孔结构的中空纤维为原料,搭建三维气凝胶网络,利用气凝胶的介孔结构和纤维的微孔结构,分别负载不同药物,实现药物的阶梯状分类释放。
本发明的目的通过下述技术方案予以实现。
三维网状埃洛石气凝胶材料及其制备方法,按照下述步骤进行:
步骤1,将0.1-20重量份埃洛石纳米管加入到50重量份去离子水和50重量份乙醇的混合液中,超声分散均匀,得到埃洛石分散液;将0.1-20重量份苯乙烯磺酸钠,0.01-2重量份聚二乙烯基硅氧烷,0.01-0.5重量份引发剂,0.05-10重量份呋塞米加入到50重量份去离子水和50重量份乙醇的混合液中,搅拌后将上述溶液加入到埃洛石分散液中,超声分散均匀,抽真空后保持真空,恢复到常压,重复抽真空三次后,洗涤,得到步骤1产物;
聚二乙烯基硅氧烷为数均分子量500-5000,优选1000—3000,乙烯基含量摩尔百分数(即乙烯基摩尔数与整个氨基单封端的聚二甲基二乙烯基硅氧烷摩尔数的比例)0.1-5%的氨基单封端的聚二甲基二乙烯基硅氧烷或者氨基双封端的聚二甲基二乙烯基硅氧烷,购自美国道康宁公司。
步骤2,将步骤1产物分散于100重量份水中,水浴升温至50-80℃引发聚合反应,聚合反应时间至少为48h,得到步骤2产物;
步骤3,将步骤2产物洗涤后分散于100重量份水中,加入0.05-20重量份的硝苯砒碇和0.01-2重量份引发剂,搅拌均匀后,水浴升温至60-100℃引发聚合反应,聚合反应时间至少为36h,制得三维网状埃洛石水凝胶;
步骤4,将三维网状埃洛石水凝胶置于co2超临界高压萃取装置中,以co2为介质在温度10-300℃和气压1-20mpa下进行超临界干燥至少1h,可得到三维网状埃洛石气凝胶材料。
引发剂选择过氧化二苯甲酰(bpo)或者偶氮二异丁腈(abin)。
在步骤1中,埃洛石纳米管为1-10重量份,加入到去离子水和乙醇的混合液中超声分散1h,苯乙烯磺酸钠为1-10重量份,聚二乙烯基硅氧烷为0.1-1重量份,引发剂为0.01-0.1重量份,呋塞米为0.1-5重量份,加入到去离子水和乙醇的混合液中搅拌10-60min后,加入到埃洛石分散液中,超声分散30min,将上述混合液抽真空后保持1h。
在步骤2中,在进行聚合反应时选择在50-80℃下聚合30-60min,然后降温至10-60℃下聚合12-24h,再依次在80℃,90℃和100℃下分别聚合2-8h。
在步骤3中,向上述分散液中加入0.1-15重量份硝苯砒碇和0.01-1重量份引发剂搅拌1h。
在步骤3中,在进行聚合反应时选择在60-100℃下聚合20-50min,然后降温至30-70℃下聚合20-30h,再依次在80℃,90℃和100℃下分别聚合2h。
在步骤4中,进行超临界干燥的时间为2-4h,优选3h。
埃洛石纳米管管壁内侧带有正电荷,管壁外侧带有负电荷,步骤1中加入的苯乙烯磺酸钠带有负电荷,苯乙烯磺酸钠通过静电作用吸附在埃洛石纳米管内壁上,同时在步骤1中加入的聚二乙烯基硅氧烷、引发剂和呋塞米也分散至埃洛石纳米管中空结构中,埃洛石纳米管中空结构为三维网状埃洛石气凝胶材料提供微孔结构,在经过抽真空、洗涤,在步骤2中聚二乙烯基硅氧烷与苯乙烯磺酸钠发生共聚,形成埃洛石纳米管内部形成交联结构,将呋塞米负载在埃洛石纳米管内,步骤3使得位于埃洛石纳米管中空结构外的聚二乙烯基硅氧烷上的乙烯基功能团之间在引发剂的作用下发生聚合,以使埃洛石纳米管和聚二乙烯基硅氧烷共同形成三维网状结构,上述三维网状结构为三维网状埃洛石气凝胶材料提供介孔结构,同时将硝苯砒碇负载在介孔结构中。
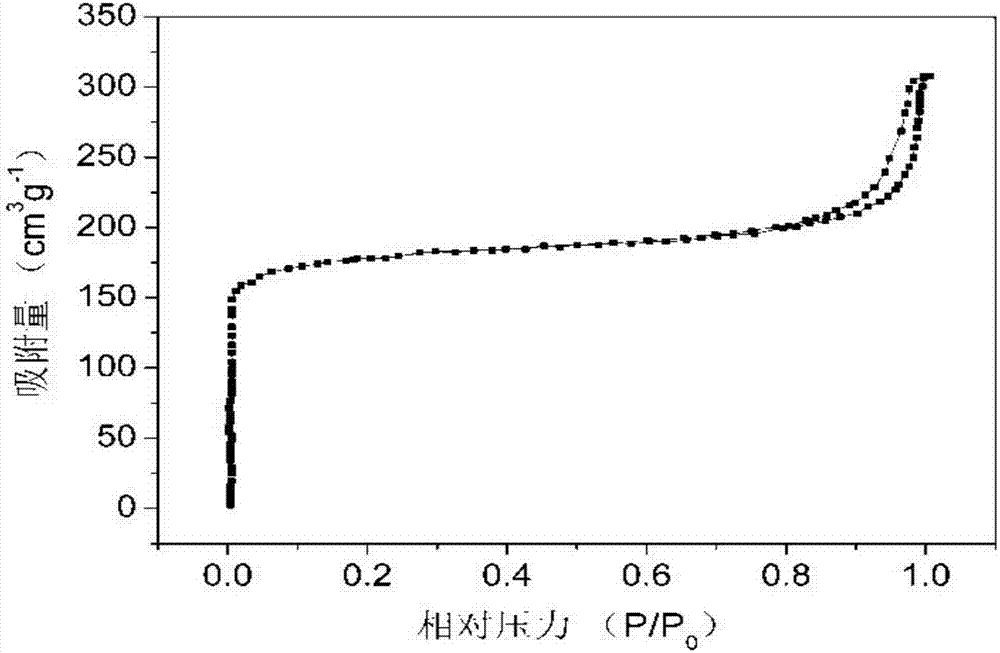
采用贝士德仪器科技(北京)有限公司3h-2000ps1型静态容量法比表面积及孔径测试仪分析根据本发明所述方法制备的复合材料的n2吸附-脱附曲线,如图1。从图中可以看出,该材料的n2吸附-脱附曲线为iupac分类中h1型迟滞环的iv类等温曲线,即由介孔结构产生。说明材料本身具有介观尺度的孔结构。从低压端点分布出现垂直上升趋势,可以看出样品内部存在较多微孔,是由于微孔内部强吸附势造成的。由氮气吸附脱附等温线数据,该样品比表面积可达到602.14m2g-1,该材料内部同时存在介孔-微孔二级孔结构,经多组测量平均比表面积可达600-608m2g-1。
将n2吸附-脱附曲线中的数据,通过bjh公式和开尔文公式,代入相关数值,可整理得孔径的计算方程rk=-0.943/ln(p/p0),单位为nm,同时再加上吸附层厚度t=0.384[-5/ln(p/p0)]^(1/3),可得实际孔径为r=rk+t,故孔径是受相对压力影响的函数,这样一来可以求得在不同的相对压力下的孔径,可以计算求得材料中存在两个孔径尖分布,一种为10.55nm,另一种为20.58μm,经多组测量纳米尺度孔隙平均可达10-12nm,微米尺度孔隙平均可达20-22μm。由此可见,材料同时存在纳米尺度和微米尺度孔隙。
采用荷兰philips的nanosem430场发射扫描电子显微镜对利用本发明所述方法制备的复合材料的微观形貌进行观察,如图2所示。从图中可以看出,埃洛石纳米管纤维成功构建为网络孔隙结构,孔径尺寸在介孔尺度。与埃洛石纳米管内壁微孔结构结合,实现了双重负载体系的构建。
参照文献(李德贵,纳米纤维素基药物缓释材料的制备及表征,华南理工大学,2016)中所述方法对本发明制备的材料进行缓释性能测试表征。分别对两个缓释台阶的缓释产物进行红外光谱检测,结果如图3所示,通过与标准图谱对照,证实率先释放的是硝苯吡啶,随后释放的是呋塞米,实现了不同药物的多尺度负载与多次释放。
将利用本发明所述方法制备的复合材料负载药物后置于模拟人体消化液中,测定其药物释放效果,从图4中可以看出,随着浸泡时间的逐渐增加,负载于介孔中的药物硝苯砒碇率先释放,随着浸泡时间的进一步增加,负载于微孔中的呋塞米药物后续释放,实现了一次服药,多次给药的效果。
附图说明
图1是三维网状埃洛石气凝胶材料的n2吸附-脱附曲线;
图2是三维网状埃洛石气凝胶材料的电镜照片;
图3是三维网状埃洛石气凝胶材料的释放产物测试曲线;
图4是三维网状埃洛石气凝胶材料的负载药物释放曲线。
具体实施方式
下面通过具体的实施例对本发明的技术方案作进一步的说明。
实施例1.
将5重量份埃洛石纳米管加入到50重量份去离子水和50重量份乙醇的混合液中,超声分散1h,得到埃洛石分散液,将10重量份苯乙烯磺酸钠,0.3重量份聚二乙烯基硅氧烷(数均分子量5000,乙烯基含量摩尔百分数0.1%的氨基单封端的聚二甲基二乙烯基硅氧烷),0.07重量份氧化二苯甲酰(bpo),3重量份呋塞米加入到50重量份去离子水和50重量份乙醇的混合液中,搅拌50min,加入到埃洛石分散液中,超声分散30min,将上述混合液抽真空后保持1h,随后恢复到常压,重复抽真空步骤三次后,将产物洗涤,然后分散于100重量份水中,置于70℃的水浴条件下预聚合24min后在20℃的恒温水浴中聚合19小时,随后将其依次在80℃,90℃,100℃的恒温水浴中分别聚合3h,将产物洗涤后分散于100重量份水中,加入9重量份的硝苯砒碇和0.3重量份氧化二苯甲酰(bpo),搅拌1h后置于100℃的水浴条件下预聚合40min,然后在56℃的恒温水浴中聚合24小时,随后将其依次在80℃,90℃,100℃的恒温水浴中分别聚合2h后制得三维网状埃洛石水凝胶,将产物置于co2超临界高压萃取装置中,以co2为介质在温度20℃和气压12mpa下进行超临界干燥3h,可得到三维网状埃洛石气凝胶材料。
实施例2.
将7重量份埃洛石纳米管加入到50重量份去离子水和50重量份乙醇的混合液中,超声分散1h,得到埃洛石分散液,将1重量份苯乙烯磺酸钠,0.5重量份聚二乙烯基硅氧烷(数均分子量500,乙烯基含量摩尔百分数5%的氨基双封端的聚二甲基二乙烯基硅氧烷),0.01重量份偶氮二异丁腈(abin),5重量份呋塞米加入到50重量份去离子水和50重量份乙醇的混合液中,搅拌11min,加入到埃洛石分散液中,超声分散30min,将上述混合液抽真空后保持1h,随后恢复到常压,重复抽真空步骤三次后,将产物洗涤,然后分散于100重量份水中,置于75℃的水浴条件下预聚合35min后在60℃的恒温水浴中聚合12小时,随后将其依次在80℃,90℃,100℃的恒温水浴中分别聚合5h,将产物洗涤后分散于100重量份水中,加入5重量份的硝苯砒碇和0.5重量份偶氮二异丁腈(abin),搅拌1h后置于85℃的水浴条件下预聚合48min,然后在40℃的恒温水浴中聚合28小时,随后将其依次在80℃,90℃,100℃的恒温水浴中分别聚合2h后制得三维网状埃洛石水凝胶,将产物置于co2超临界高压萃取装置中,以co2为介质在温度10℃和气压14mpa下进行超临界干燥3h,可得到三维网状埃洛石气凝胶材料。
实施例3.
将9重量份埃洛石纳米管加入到50重量份去离子水和50重量份乙醇的混合液中,超声分散1h,得到埃洛石分散液,将4重量份苯乙烯磺酸钠,0.8重量份聚二乙烯基硅氧烷(数均分子量1000,乙烯基含量摩尔百分数4%的氨基双封端的聚二甲基二乙烯基硅氧烷),0.1重量份偶氮二异丁腈(abin),0.7重量份呋塞米加入到50重量份去离子水和50重量份乙醇的混合液中,搅拌13min,加入到埃洛石分散液中,超声分散30min,将上述混合液抽真空后保持1h,随后恢复到常压,重复抽真空步骤三次后,将产物洗涤,然后分散于100重量份水中,置于50℃的水浴条件下预聚合30min后在10℃的恒温水浴中聚合18小时,随后将其依次在80℃,90℃,100℃的恒温水浴中分别聚合8h,将产物洗涤后分散于100重量份水中,加入0.1重量份的硝苯砒碇和0.7重量份偶氮二异丁腈(abin),搅拌1h后置于90℃的水浴条件下预聚合35min,然后在30℃的恒温水浴中聚合30小时,随后将其依次在80℃,90℃,100℃的恒温水浴中分别聚合2h后制得三维网状埃洛石水凝胶,将产物置于co2超临界高压萃取装置中,以co2为介质在温度100℃和气压3mpa下进行超临界干燥3h,可得到三维网状埃洛石气凝胶材料。
实施例4.
将1重量份埃洛石纳米管加入到50重量份去离子水和50重量份乙醇的混合液中,超声分散1h,得到埃洛石分散液,将6重量份苯乙烯磺酸钠,0.7重量份聚二乙烯基硅氧烷(数均分子量3000,乙烯基含量摩尔百分数1%的氨基单封端的聚二甲基二乙烯基硅氧烷),0.03重量份氧化二苯甲酰(bpo),0.1重量份呋塞米加入到50重量份去离子水和50重量份乙醇的混合液中,搅拌20min,加入到埃洛石分散液中,超声分散30min,将上述混合液抽真空后保持1h,随后恢复到常压,重复抽真空步骤三次后,将产物洗涤,然后分散于100重量份水中,置于55℃的水浴条件下预聚合60min后在55℃的恒温水浴中聚合24小时,随后将其依次在80℃,90℃,100℃的恒温水浴中分别聚合2h,将产物洗涤后分散于100重量份水中,加入4重量份的硝苯砒碇和0.8重量份氧化二苯甲酰(bpo),搅拌1h后置于80℃的水浴条件下预聚合39min,然后在37℃的恒温水浴中聚合20小时,随后将其依次在80℃,90℃,100℃的恒温水浴中分别聚合2h后制得三维网状埃洛石水凝胶,将产物置于co2超临界高压萃取装置中,以co2为介质在温度300℃和气压5mpa下进行超临界干燥3h,可得到三维网状埃洛石气凝胶材料。
实施例5.
将3重量份埃洛石纳米管加入到50重量份去离子水和50重量份乙醇的混合液中,超声分散1h,得到埃洛石分散液,将2重量份苯乙烯磺酸钠,1重量份聚二乙烯基硅氧烷(数均分子量2500,乙烯基含量摩尔百分数3%的氨基双封端的聚二甲基二乙烯基硅氧烷),0.02重量份氧化二苯甲酰(bpo),0.8重量份呋塞米加入到50重量份去离子水和50重量份乙醇的混合液中,搅拌60min,加入到埃洛石分散液中,超声分散30min,将上述混合液抽真空后保持1h,随后恢复到常压,重复抽真空步骤三次后,将产物洗涤,然后分散于100重量份水中,置于60℃的水浴条件下预聚合20min后在19℃的恒温水浴中聚合15小时,随后将其依次在80℃,90℃,100℃的恒温水浴中分别聚合6h,将产物洗涤后分散于100重量份水中,加入15重量份的硝苯砒碇和0.01重量份氧化二苯甲酰(bpo),搅拌1h后置于60℃的水浴条件下预聚合20min,然后在70℃的恒温水浴中聚合27小时,随后将其依次在80℃,90℃,100℃的恒温水浴中分别聚合2h后制得三维网状埃洛石水凝胶,将产物置于co2超临界高压萃取装置中,以co2为介质在温度150℃和气压20mpa下进行超临界干燥3h,可得到三维网状埃洛石气凝胶材料。
实施例6.
将10重量份埃洛石纳米管加入到50重量份去离子水和50重量份乙醇的混合液中,超声分散1h,得到埃洛石分散液,将8重量份苯乙烯磺酸钠,0.1重量份聚二乙烯基硅氧烷(数均分子量4000,乙烯基含量摩尔百分数0.5%的氨基双封端的聚二甲基二乙烯基硅氧烷),0.05重量份偶氮二异丁腈(abin),0.3重量份呋塞米加入到50重量份去离子水和50重量份乙醇的混合液中,搅拌10min,加入到埃洛石分散液中,超声分散30min,将上述混合液抽真空后保持1h,随后恢复到常压,重复抽真空步骤三次后,将产物洗涤,然后分散于100重量份水中,置于80℃的水浴条件下预聚合37min后在50℃的恒温水浴中聚合20小时,随后将其依次在80℃,90℃,100℃的恒温水浴中分别聚合4h,将产物洗涤后分散于100重量份水中,加入6重量份的硝苯砒碇和1重量份偶氮二异丁腈(abin),搅拌1h后置于75℃的水浴条件下预聚合50min,然后在45℃的恒温水浴中聚合25小时,随后将其依次在80℃,90℃,100℃的恒温水浴中分别聚合2h后制得三维网状埃洛石水凝胶,将产物置于co2超临界高压萃取装置中,以co2为介质在温度55℃和气压1mpa下进行超临界干燥3h,可得到三维网状埃洛石气凝胶材料。
实施例7
将0.1重量份埃洛石纳米管加入到50重量份去离子水和50重量份乙醇的混合液中,超声分散1h,得到埃洛石分散液,将0.1重量份苯乙烯磺酸钠,0.01重量份聚二乙烯基硅氧烷(数均分子量3500,乙烯基含量摩尔百分数3%的氨基单封端的聚二甲基二乙烯基硅氧烷),0.01重量份偶氮二异丁腈(abin),0.05重量份呋塞米加入到50重量份去离子水和50重量份乙醇的混合液中,搅拌11min,加入到埃洛石分散液中,超声分散30min,将上述混合液抽真空后保持1h,随后恢复到常压,重复抽真空步骤三次后,将产物洗涤,然后分散于100重量份水中,置于75℃的水浴条件下预聚合35min后在60℃的恒温水浴中聚合12小时,随后将其依次在80℃,90℃,100℃的恒温水浴中分别聚合5h,将产物洗涤后分散于100重量份水中,加入0.05重量份的硝苯砒碇和0.01重量份偶氮二异丁腈(abin),搅拌1h后置于85℃的水浴条件下预聚合48min,然后在40℃的恒温水浴中聚合28小时,随后将其依次在80℃,90℃,100℃的恒温水浴中分别聚合2h后制得三维网状埃洛石水凝胶,将产物置于co2超临界高压萃取装置中,以co2为介质在温度50℃和气压20mpa下进行超临界干燥2h,可得到三维网状埃洛石气凝胶材料。
实施例8
将20重量份埃洛石纳米管加入到50重量份去离子水和50重量份乙醇的混合液中,超声分散1h,得到埃洛石分散液,将20重量份苯乙烯磺酸钠,2重量份聚二乙烯基硅氧烷(数均分子量1500,乙烯基含量摩尔百分数4.5%的氨基单封端的聚二甲基二乙烯基硅氧烷),0.5重量份氧化二苯甲酰(bpo),10重量份呋塞米加入到50重量份去离子水和50重量份乙醇的混合液中,搅拌60min,加入到埃洛石分散液中,超声分散30min,将上述混合液抽真空后保持1h,随后恢复到常压,重复抽真空步骤三次后,将产物洗涤,然后分散于100重量份水中,置于60℃的水浴条件下预聚合20min后在19℃的恒温水浴中聚合15小时,随后将其依次在80℃,90℃,100℃的恒温水浴中分别聚合6h,将产物洗涤后分散于100重量份水中,加入20重量份的硝苯砒碇和2重量份氧化二苯甲酰(bpo),搅拌1h后置于60℃的水浴条件下预聚合20min,然后在70℃的恒温水浴中聚合27小时,随后将其依次在80℃,90℃,100℃的恒温水浴中分别聚合2h后制得三维网状埃洛石水凝胶,将产物置于co2超临界高压萃取装置中,以co2为介质在温度120℃和气压13mpa下进行超临界干燥4h,可得到三维网状埃洛石气凝胶材料。
以上对本发明做了示例性的描述,应该说明的是,在不脱离本发明的核心的情况下,任何简单的变形、修改或者其他本领域技术人员能够不花费创造性劳动的等同替换均落入本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!