一种不使用碘化亚铜的他扎罗汀的制备方法与流程
本发明涉及一种药物合成方法,特别涉及不使用碘化亚铜的他扎罗汀的制备方法。
背景技术:
他扎罗汀(i)是第一个受体选择性、第三代芳香维a酸类药物,主要选择性地结合二种维a酸受体(rar-β;rar-γ),但不与维a酸x受体(rxr)结合。临床安全、有效地用于治疗银屑病、痤疮,并用于角化异常性疾病、毛囊皮脂腺疾病、皮肤癌前期病变。
他扎罗汀的合成方法有许多报道,但各具有一些缺点,不适于工业化生产。
如美国专利1992年us5089509,以及1997us5602130均公开了他扎罗汀的不同合成方法。上述两种方法的最后一步合成均是用4、4-二甲基苯并噻喃-6-基-乙炔与6-氯代烟酸乙酯在有正丁基锂、膦钯配合物、氯化锌存在的条件下进行反应,得到终产物他扎罗汀。因为两种方法都要使用钯复合物作催化剂,不仅价格昂贵,而且重金属的回收非常繁琐,回收成本高。另外,由于反应体系中使用了正丁基锂,需无水反应,反应条件较苛刻。
2006年美国专利us7273937报道的他扎罗汀的合成,其最后一步合成的操作方法需要低温,且需要过柱,这些复杂的手段都不利于工业化规模生产。
2009年专利wo2009/116075a2提供了一种他扎罗汀的合成方法。其合成线路如下:
最后一步合成、后处理及产物他扎罗汀的纯化方法如下:
将6-氯烟酸乙酯(该专利文献图7)溶解于甲苯中,然后加入碳酸钾、三苯基膦、5%钯炭,在45~50℃左右搅拌2h,然后加入4、4-二甲基苯并噻喃-6-基-乙炔(该专利文献图6)和碘化亚铜,然后升温至105~115℃左右反应10h。反应结束后降至室温,过滤,水稀释,搅拌反应0.5h,分液;水层用甲苯提取,合并后的有机层用无水硫酸镁干燥,过滤,有机层减压浓缩,加入正己烷降至5~-10℃析晶1h,过滤干燥得到他扎罗汀。单步收率60.0%。
该步反应机理如下:
有碘化亚铜作共催化剂的sonogashira反应常被认为发生了双环反应。钯催化剂被氧化加成上r1-x(r1=芳基、杂环基团、烯烃:x=碘、溴、氯、氟),产生具有催化活性的钯化合物。下面一步就是钯催化的环与亚铜做共催化剂的环相结合继续反应,反应速率控制步是在铜环下产生的铜炔能否与钯催化剂的中间体生成r1pd(-c=cr2)l2物质反应,这个是由铜炔在产生的催化剂条件下通过顺反异构和还原消除而得到的。
虽然该方法较前几种方法经济,但是存在以下问题:
1、最后一步反应用到了催化剂碘化亚铜
碘化亚铜容易与产品形成络合物,在产品中不易除去,造成产品重金属含量超标,无法达到药品的质量标准。
此外,合成过程中产生的废水中含重金属,亦会对环境造成影响;
2、反应在高温条件下进行较长时间(105~115℃左右反应10h),不仅耗能较高,更主要的是,极易造成产品中杂质含量增大,影响产品的后续处理,并造成产品收率的降低。
技术实现要素:
本发明的目的在于克服wo2009/116075a2的缺点,提供一种无碘化亚铜作共催化剂,催化剂容易回收,对环境友好,反应时间短,产品收率高,纯度高的他扎罗汀制备方法。
本发明对上述专利方法中的催化剂进行了改进,技术方案为:
a不使用碘化亚铜;
b反应的步骤是:
1)先将钯催化剂、三苯基膦加入特定有机溶剂中,搅拌,通氮置换;
2)再加入4、4-二甲基苯并噻喃-6-基-乙炔、6-氯代烟酸乙酯和缚酸剂进行反应;
c反应温度为50~100℃;
d反应时间为3~7h。
产物的后处理方法是:过滤回收催化剂,分液,提取水层,碱洗有机层,干燥,浓缩,加入有机溶剂析晶,过滤分离,得到他扎罗汀。
回收的催化剂钯炭可以重新用于上述反应。
所述特定有机溶剂选自n-甲基吡咯烷酮、dmf、甲苯中的一种或几种。
所述缚酸剂选自碳酸钾、碳酸钠、碳酸氢钠、乙酸钾的一种或几种。
所述催化剂为钯炭。
所述的反应温度为50~100℃。
所述的反应时间为3~7h。
析晶所用有机溶剂选自乙酸乙酯、正己烷、丙酮中的一种或几种。
析晶温度为5~-15℃,其优选温度为0~-10℃。
析晶时间为1~6h,其优选时间为1~2h。
本发明的反应机理如下:
在此反应中,钯催化剂被氧化加成上r1-x(r1=芳基、杂环基团、烯烃:x=碘、溴、氯、氟)产生具有催化活性的钯化合物,然后通过磷配合物与具有催化活性的钯化合物形成稳定的六面体结构,在缚酸剂的作用下与炔化物直接反应,脱去生成的酸,生成产品,钯催化剂恢复催化活性,继续参与反应。由于此反应无铜离子参与反应不会影响产品质量。
本发明的有益效果:
1、不使用碘化亚铜
碘化亚铜容易与产品形成络合物,在产品中不易除去,造成产品重金属含量超标,无法达到药品的质量标准。本方法不产生此问题。
2、催化剂容易回收
用过滤方式就可以回收催化剂。回收后的催化剂经水洗,干燥后可以重新使用(见实施例4)。wo2009/116075a2中使用到了碘化亚铜,钯炭与碘离子形成了络合物,无法直接回收使用。
3、对环境友好
反应结束后,钯炭被过滤回收,后处理中产生的废水无重金属,不会对环境产生重金属污染。
4、比现有技术反应时间短
wo2009/116075a2在105~115℃左右反应10h,本方法60~100℃反应3~7h。
5、产品收率高,纯度高
因为反应时间相比较短,产生杂质机会相对较小,产物纯度高,后续处理简单,产物损失少,因此收率高(80%以上,比报道的文献高得多,此步收率高近20个百分点)。
附图说明
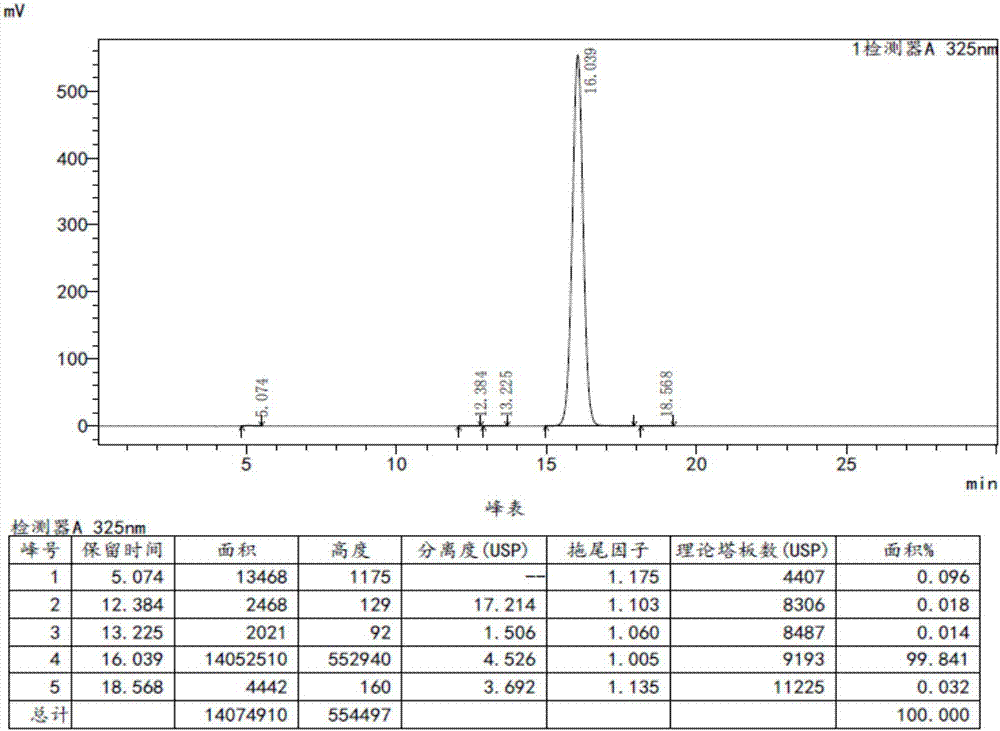
图1是实施例1所得产物他扎罗汀的hplc检测图。
图2是实施例2所得产物他扎罗汀的hplc检测图。
图3是实施例3所得产物他扎罗汀的hplc检测图。
具体实施方式
以下实施例中提到的各原料、试剂均从市售渠道获得。制得的他扎罗汀用高效液相色谱检测。
色谱条件是:色谱柱为shimadzuvp-ods,150×4.6mm,5μm;流动相为异丙醇―乙腈―水(38:27:35);检测波长为325nm;流速为0.5ml/min;进样量为20μl。
实施例1
洁净的250ml三口瓶中加入dmf62g、10%钯碳0.81g、三苯基膦0.40g,搅拌,通氮置换。保持微弱氮气流,搅拌10~20分钟。依次加入4、4-二甲基苯并噻喃-6-基-乙炔15.0g和6-氯代烟酸乙酯16.5g、乙酸钾12.5g、纯化水2.43g、dmf25g。升温至85℃,反应4小时,然后取样tlc监测,直至4、4-二甲基苯并噻喃-6-基-乙炔斑点消失(展开剂二氯甲烷:正己烷=1:6)。
降温至室温,抽滤,滤液中加二氯甲烷55g,饮用水470g,搅拌,静置,分液。合并有机相,用饱和碳酸氢钠溶液250g分两次洗涤。分液,合并有机相,用饱和氯化钠溶液250g分两次洗涤。分液,合并有机相,加入无水硫酸镁20g搅拌干燥。过滤,滤液减压浓缩至干。加入乙酸乙酯15g,升温回流10~20分钟,物料溶清后加入正己烷15g,降温至-7℃,析晶1h。抽滤,在60℃减压干燥3h,得他扎罗汀21.1g,收率为80.9%。
实施例2
洁净的250ml三口瓶中加入n-甲基吡咯烷酮100g、10%钯碳0.81g、三苯基膦0.80g,搅拌,通氮置换。保持微弱氮气流,搅拌10~20分钟。依次加入4、4-二甲基苯并噻喃-6-基-乙炔15.0g和6-氯代烟酸乙酯18.0g、碳酸钾12.5g、纯化水2.43g。升温至90℃,反应5小时,然后取样tlc监测,直至4、4-二甲基苯并噻喃-6-基-乙炔斑点消失(展开剂二氯甲烷:正己烷=1:6)。
降温至室温,抽滤,滤液中加二氯甲烷55g,饮用水470g,搅拌,静置,分液。合并有机相,用10%碳酸钠溶液250g分两次洗涤。分液,合并有机相,用饱和氯化钠溶液250g分两次洗涤。分液,合并有机相,加入无水硫酸镁20g搅拌干燥。过滤,滤液减压浓缩至干。加入乙酸乙酯15g,升温回流10~20分钟,物料溶清后加入正己烷30g,降温至0℃,析晶1h。抽滤,在60℃减压干燥3h,得他扎罗汀22.1g,收率为84.8%。
实施例3
洁净的250ml三口瓶中加入甲苯100g、10%钯碳0.81g、三苯基膦0.40g,搅拌,通氮置换。保持微弱氮气流,搅拌10~20分钟。依次加入4、4-二甲基苯并噻喃-6-基-乙炔15.0g和6-氯代烟酸乙酯15.0g、乙酸钾15.0g、纯化水1.62g。升温至95℃,反应6小时,然后取样tlc监测,直至4、4-二甲基苯并噻喃-6-基-乙炔斑点消失(展开剂二氯甲烷:正己烷=1:6)。
降温至室温,抽滤,滤液中加二氯甲烷55g,饮用水470g,搅拌,静置,分液。合并有机相,用10%碳酸钠溶液250g分两次洗涤。分液,合并有机相,用饱和氯化钠溶液250g分两次洗涤。分液,合并有机相,加入无水硫酸镁20g搅拌干燥。过滤,滤液减压浓缩至干。加入乙酸乙酯15g,升温回流10~20分钟,物料溶清后加入正己烷30g,降温至5℃,析晶1h。抽滤,在60℃减压干燥3h,得他扎罗汀21.6g,收率为82.8%。
实施例4
洁净的250ml三口瓶中加入dmf90g、回收钯碳0.81g、三苯基膦0.40g,搅拌,通氮置换。保持微弱氮气流,搅拌10~20分钟。依次加入4、4-二甲基苯并噻喃-6-基-乙炔15.0g和6-氯代烟酸乙酯15.0g、乙酸钾15.0g、纯化水1.62g。升温至80℃,反应5小时,然后取样tlc监测,直至4、4-二甲基苯并噻喃-6-基-乙炔斑点消失(展开剂二氯甲烷:正己烷=1:6)。降温至室温,抽滤,滤液中加二氯甲烷55g,饮用水470g,搅拌,静置,分液。合并有机相,用饱和碳酸氢钠溶液250g分两次洗涤。分液,合并有机相,用饱和氯化钠溶液250g分两次洗涤。分液,合并有机相,加入无水硫酸镁20g搅拌干燥。过滤,滤液减压浓缩至干。加入乙酸乙酯15g,升温回流10~20分钟,物料溶清后加入正己烷15g,降温至-5℃,析晶1h。抽滤,在60℃减压干燥3h,得他扎罗汀22.5g,收率为86.3%。
- 还没有人留言评论。精彩留言会获得点赞!