磷酸泰地唑胺铵盐及其晶型、制备方法和医药用途与流程
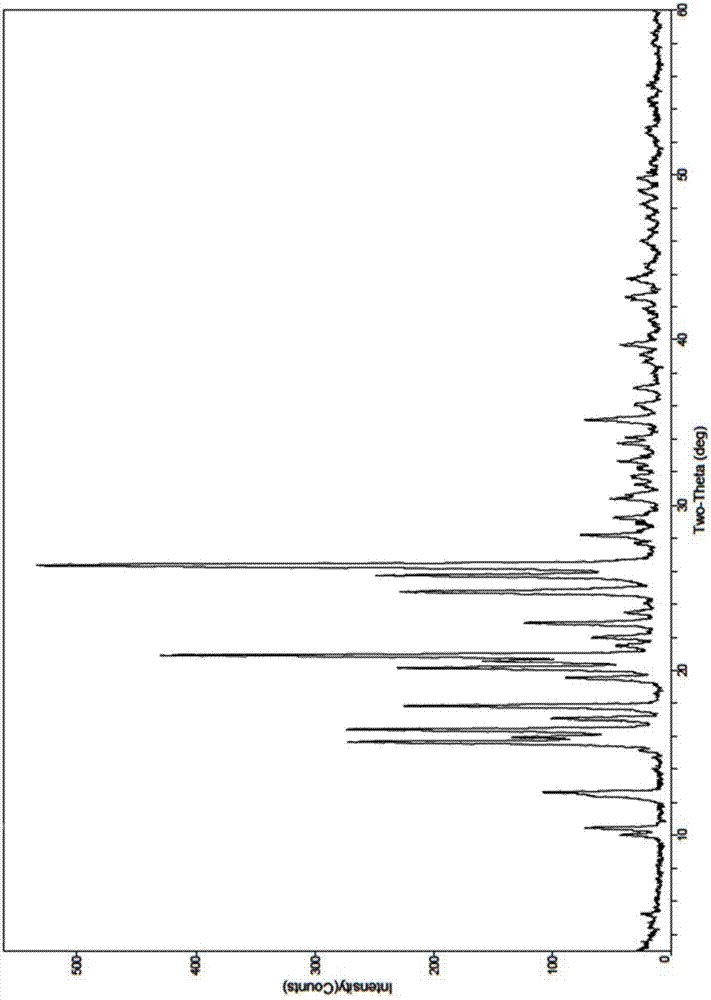
本发明涉及化学制药领域,更具体的说,本发明涉及磷酸泰地唑胺铵盐及其晶型、磷酸泰地唑胺铵盐及其晶型的制备方法、以及磷酸泰地唑胺铵盐及其晶型的医药用途。技术背景磷酸泰地唑胺是一种新的恶唑烷酮类抗菌素,由cubist制药公司开发,于2014年6月20日获美国fda批准上市。用于治疗金黄色葡萄球菌(包括耐甲氧西林菌株和甲氧西林敏感菌株)、各种链球菌及肠球菌等革兰阳性细菌引起的成人急性细菌性皮肤和皮肤组织感染。这是fda继5月23日批准达巴万星之后批准用于治疗absssi感染的第二个抗菌素新药。国内由拜耳医药保健有限公司在2014年6月申报注射用粉针的进口临床。磷酸泰地唑胺是一种前药,是细菌蛋白合成抑制剂,在体内可被磷酸酶迅速转化为具有生物活性的泰地唑胺。后者能够和细菌的核糖体50s亚基结合,干扰包括mrna、30s核糖体及起始因子等的70s起始复合物的形成,从而抑制细菌蛋白的合成。由于此抑制过程处于蛋白翻译的早期阶段,因此不易与其它机理的抗生素发生交叉耐药。尽管自2000年辉瑞的同类抗菌素利奈唑胺获得美国食品药品监督局(fda)批准以后,至少有10个同类化合物进入临床,但磷酸泰地唑胺是第一个获得fda批准的二代恶唑烷酮类抗生素。和一代产品利奈唑胺相比,磷酸泰地唑胺对一些细菌的体外抑制活性要高2-8倍,安全性在一定程度上也有所提高。专利文献cn1894242和cn101982468分别公开了磷酸泰地唑胺的化合物制备方法,专利文献us20100227839a1、wo2015158202a1分别公开了磷酸泰地唑胺的相关晶型。对于多晶型药物而言,不同的晶型可以具有不同的物理化学性质,包括熔点、化学稳定性、表观溶解度、溶解速率、光学和机械性质等,而这些物化性能直接决定了某特定的晶型是否可以成药,并且它们影响到原料药和制剂的质量。因此,尽管现有技术已经披露了磷酸泰地唑胺及其晶型,但是磷酸泰地唑胺及其晶型的溶解性差,通常采用反应结晶方式除杂,其杂质去除效果不理想,颗粒度小,不利于过滤烘干,同时其澄清度不易达标。因此有必要开发理化性质优良的、有利于工业化生产的、结晶效果良好的磷酸泰地唑胺盐及其晶型来满足日益严苛的药物质量要求。技术实现要素:本发明涉及磷酸泰地唑胺铵盐,化学名为:(r)-3-(4-(2-(2-甲基四唑-5-基)吡啶-5-基)-3-氟苯基)-5-羟甲基噁唑烷-2-酮磷酸铵,其结构式如下所示:本发明涉及磷酸泰地唑胺铵盐制备方法,其制备方法采用以下合成路线:磷酸泰地唑胺(式ⅱ)与氨水反应即可得到磷酸泰地唑胺铵盐(式ⅰ)。本发明还涉及一种化学和物理稳定性均较好的磷酸泰地唑胺铵盐的晶型a,所述晶型在化学稳定性和加工(过滤和干燥)适应性等方面具有优异的性质。本发明所述的磷酸泰地唑胺铵盐晶型a的x-射线粉末衍射(xrd)图在以下衍射角2θ处有特征峰:15.7±0.2°、16.4±0.2°、17.8±0.2°、20.1±0.2°、20.9±0.2°、24.7±0.2°、25.7±0.2°、26.4±0.2°。优选地,本发明所述的磷酸泰地唑胺铵盐晶型a的x-射线粉末衍射图进一步在以下2θ处有特征峰:10.4±0.2°、12.6±0.2°、16.0±0.2°、17.1±0.2°、19.5±0.2°、20.6±0.2°、22.9±0.2°。优选地,本发明所述的磷酸泰地唑胺铵盐晶型a的x-射线粉末衍射图再进一步在以下2θ处有特征峰:10.0±0.2°、21.5±0.2°、22.0±0.2°、23.5±0.2°、28.2±0.2°、29.2±0.2°、30.4±0.2°、32.6±0.2°、33.7±0.2°、35.2±0.2°。优选地,本发明所述的磷酸泰地唑胺铵盐晶型a的x-射线粉末衍射谱图具有如下表1所示的2θ和相对强度数据:表1更优选地,本发明所述的磷酸泰地唑胺铵盐晶型a具有如图1所示的x-射线粉末衍射谱图。此外,本发明所述的磷酸泰地唑胺铵盐晶型a,可以用kbr压片测得的红外吸收图谱表征,其在约3199.90cm-1、1740.07cm-1、1689.84cm-1、1623.86cm-1、1453.34cm-1、1417.35cm-1、1342.30cm-1、1227.48cm-1、1199.14cm-1、1152.80cm-1、1100.74cm-1、1045.63cm-1、927.48cm-1、877.23cm-1、834.00cm-1、786.16cm-1、753.33cm-1、715.95cm-1、554.91cm-1、525.97cm-1处有吸收峰。本发明所述的磷酸泰地唑胺铵盐晶型a具有如图2所示的红外谱图。本发明所述的磷酸泰地唑胺铵盐晶型a的差示扫描量热(dsc)图谱在249℃处有最大吸收峰。本发明所述的磷酸泰地唑胺铵盐晶型a具有如图3所示的dsc谱图。本发明所述的磷酸泰地唑胺铵盐晶型a具有如图4所示的tga谱图。本发明所述的磷酸泰地唑胺铵盐晶型a的粒度分布如下表2所示:表2本发明的磷酸泰地唑胺铵盐晶型a具有如图5所示的粒度分布谱图。本发明还涉及一种磷酸泰地唑胺铵盐晶型a的制备方法,该方法包括以下步骤:(1)将磷酸泰地唑胺铵盐溶液,用弱酸溶液调ph;(2)搅拌析晶;(3)过滤,得磷酸泰地唑胺铵盐晶型a。优选地,步骤(1)中所述磷酸泰地唑胺铵盐溶液可由以下方法制备:①将磷酸泰地唑胺铵盐溶于水或氨水溶液中,所述氨水溶液的质量百分浓度优选0.1%-18%,所述磷酸泰地唑胺铵盐与氨水溶液中氨水的摩尔比优选为1:1-1:15;所述磷酸泰地唑胺铵盐与水的重量比优选为1:400-1:600;或②磷酸泰地唑胺与氨水溶液反应得到,所述氨水溶液的质量百分浓度优选0.1%-25%,所述磷酸泰地唑胺与氨水溶液中氨水的摩尔比优选为1:1-1:25。优选地,步骤(1)所述弱酸为有机酸,优选为甲酸和乙酸;所述弱酸溶液的摩尔浓度为0.1-3mol/l,更优选为0.5-1mol/l;所述ph值范围为3-4。优选地,步骤(2)中所述搅拌析晶的温度为5-40℃,优选为20-30℃。本发明还涉及含有磷酸泰地唑胺铵盐及其晶型a的药物组合物,所述药物组合物包含治疗有效量的磷酸泰地唑胺铵盐或其晶型a,以及一种或多种药学上可接受的载体。所述药物组合物可为固态、半固态或液态,可制备成合适的剂型例如固体剂型,包括片剂、颗粒剂、散剂、丸剂、粉末和胶囊剂;液体剂型,包括溶液剂、糖浆剂、混悬剂、分散剂和乳剂;可注射制剂,包括溶液剂、分散剂、适于注射前在液态中溶解或悬浮的固体形式例如冻干剂。配方可适于活性成分的快速释放、持续释放或调解释放。可以是常规的、可分散的、可咀嚼的、口腔溶解的或快速熔化的制剂。给药途径包括口服、通过胃喂食管、通过十二指肠喂食管、静脉内、动脉内、肌肉内、皮下、骨内、皮肤内、阴道内、直肠内、腹膜内、透皮、鼻内、眼滴、耳滴等。所述药物组合物中药学上接受的载体或助剂,在固态制剂的情况下包括但不限于:稀释剂,例如淀粉、改性淀粉、乳糖、粉状纤维素、微晶纤维素、无水碳酸氢钙、磷酸三钙、甘露醇、山梨醇、蔗糖等;粘合剂,例如阿拉伯胶、瓜儿胶、明胶、聚乙烯吡咯烷酮、羟丙基纤维素、羟丙基甲基纤维素、聚乙二醇、共聚维酮等;崩解剂,例如淀粉、羧甲基淀粉钠、羟基乙酸淀粉钠、预胶化淀粉、交联聚维酮、交联羧甲基纤维素钠、胶体二氧化硅等;润滑剂,例如硬脂酸、硬脂酸镁、硬脂酸锌、苯甲酸钠、乙酸钠等;助流剂,例如胶体二氧化硅;复合物形成剂,例如各种级别的环糊精和树脂;释放速度控制剂,例如羟丙基纤维素、羟甲基纤维素、羟丙基甲基纤维素、乙基纤维素、甲基纤维素、甲基丙烯酸甲酯、蜡等。其他药学上可接受的载体或助剂包括但不限于成膜剂、增塑剂、着色剂、调味剂、粘度调节剂、防腐剂、抗氧化剂等。口服片剂中,通常使用的载体或助剂包括糖比如乳糖、蔗糖、甘露醇或山梨醇,纤维素制品比如玉米淀粉、小麦淀粉、明胶、甲基纤维素、羟丙基甲基纤维素、羟甲基纤维素钠和聚乙烯吡咯烷酮,还可以加入润滑剂比如硬脂酸镁、崩解剂比如交联聚乙烯吡咯烷酮,进一步地可对片剂核心进行包衣,例如形成糖皮层;口服胶囊剂中,有用的载体或助剂包括乳糖、高和低分子量聚乙二醇和干玉米淀粉;明胶胶囊剂的情况下,粉末载体例如乳糖、淀粉、纤维素衍生物、硬脂酸镁、硬脂酸与类似物;软胶囊的情况下,活性化合物可以溶解或悬浮于合适的液体中,比如脂肪油、液体石蜡或液态聚乙二醇;当以混悬液口服给药时,所述活性成分与乳化剂和悬浮剂混合;如果需要,可以加入某些甜味剂和/或调味剂和/或着色剂。每一种载体或助剂必须可接受的,能与配方中的其它成分兼容并且对于病患无害。所述药物组合物可以使用本领域技术人员公知的方法来制备。制备药物组合物时,本发明的磷酸泰地唑胺铵盐或其晶型a与一种或多种药学上可接受的载体或助溶剂相混合,任选地,与一种或多种其他的药物活性成分相混合。固态制剂可以通过直接混合、制粒等工艺来制备。液体制剂可以通过溶解、分散等工艺来制备。本发明还提供了磷酸泰地唑胺铵盐及其晶型a以及其药物组合物在制备抗感染药物中的用途,以及通过使用该药物来抗细菌感染的方法。其中,所述药物能够和细菌的核糖体50s亚基结合,从而抑制细菌蛋白的合成。前述的细菌感染,包括但不限于皮肤感染,肺炎,病毒感染后感染,腹部感染,泌尿道感染,菌血症,败血病,心内膜炎,房室间隔感染,血管穿刺感染,脑膜炎,外科手术预防,腹膜感染,骨感染,关节感染,具有甲氧西林抗性的金黄色葡萄球菌感染,具有万古霉素抗性的肠球菌感染,具有利奈唑胺抗性的有机体感染以及肺结核。本发明的发明人经过大量研究首次成功制备得到磷酸泰地唑胺铵盐及其晶型a。所述磷酸泰地唑胺铵盐的制备方法简单,有利于工业化生产;所述磷酸泰地唑胺铵盐晶型a的溶解性良好、结晶工艺简单、便于操作、污染小、可实现工业化生产;本发明磷酸泰地唑胺铵盐的晶型药物同时具备产品纯度高、理化性质优异、化学稳定性良好、加工(过滤和干燥)可再现的优点。此外,本发明的磷酸泰地唑胺铵盐晶型a与磷酸泰地唑胺的晶型相比,具有较好的溶解度、澄清度、过滤速度、吸湿性和颜色(白色)。附图说明:图1为实施例5所得磷酸泰地唑胺铵盐晶型a的x-射线粉末衍射图谱。图2为实施例5所得磷酸泰地唑胺铵盐晶型a的红外吸收光谱。图3为实施例5所得磷酸泰地唑胺铵盐晶型a的dsc图谱。图4为实施例5所得磷酸泰地唑胺铵盐晶型a的tga图谱。图5为实施例5所得磷酸泰地唑胺铵盐晶型a的粒度分布图谱。具体实施例下列实施例用于进一步解释说明本发明,但是,它们并不构成对本发明范围的限制或限定。本发明方法中所使用的磷酸泰地唑胺起始原料可以商购获得,或者按照已知方法,例如专利文献cn200480037612.2中实施例58中第二步所记载的方法制备,该文献通过引用的方式并入本申请中。本发明所使用的溶剂没有特别的限制,可采用商购的常规溶剂,例如所述氨水可以是市售氨水,包括工业氨水、分析纯氨水等。除非另有说明,本发明方法中所述的“搅拌”可以采用本领域的常规方法,例如搅拌方式包括磁力搅拌、机械搅拌,搅拌速度为50-300rpm/min,优选100-200rpm/min;本发明所述室温是指温度为18℃-30℃。本发明所涉及的x-射线粉末衍射仪器及测试条件为:x-衍射仪器型号rigakud/max-2200cu靶;扫描速度4°/min,扫描步宽0.01°。本发明所涉及的红外分光广度仪及测试条件为:红外分光光度仪型号:brwkervector22;采用kbr压片法,扫描范围400-4000cm-1。本发明涉及的dsc测试条件为:dsc检测仪型号为:netzschdsc214polyma;操作方法:升温速率10℃/min,温度范围:30-265℃。本发明涉及的tga测试条件为:tga检测仪型号为:perkinelmertga400升温速率10℃/min,温度范围:30-300℃。本发明涉及的质谱测试仪型号:agilent-1100-lcq-advantage。本发明涉及的核磁测试条件为:核磁检测仪型号为:brukeravance400;dmso,400mhz。本发明涉及的粒度分布测试条件为:粒度检测仪型号为:bt-2001激光粒度分布仪;操作方法:进行与仪器生产商的说明一致的取样说明,通过悬浮于3-4ml正庚烷中并超声2min制备得到测试样品。本发明所涉及的澄清度测试条件为:澄清度仪型号为:merckturbiquant1500ir;操作方法:取1g磷酸泰地唑胺铵盐加入容量瓶中,加入碳酸钠溶液,使钠离子与磷酸泰地唑胺的摩尔比为2:1,将溶液定容至20ml。本发明所涉及的溶解度测试条件为:shimadzu日本岛津的紫外可见分光光度计型号:uv-2660。本发明涉及的液相测试条件为:色谱柱为c18250×4.6mm,5μm;缓冲液:20mmolnh4h2po4,二乙胺调ph为6.5;流动相a:缓冲液:乙腈=85:15;流动相b:缓冲液:乙腈=20:80;检测波长:300nm;流速:1ml/min;进样量:10μl;柱温:50℃,液相条件如表3所示:表3t(min)a(%)b(%)01000358020500100600100611000681000应当强调的是,本发明技术方案中所涉及的数值或数值端点,其含义或意义的保护范围并不局限于该数字本身,本领域技术人员能够理解,它们包含了那些已被本领域广为接受的可允许误差范围,例如实验误差、测量误差、统计误差和随机误差等等,而这些误差范围均包含在本发明的范围之内。实施例1-18所用的磷酸泰地唑胺粗品是按照专利文献cn200480037612.2中实施例58中第二步中所记载的方法制备。实施例1将磷酸泰地唑胺粗品5g(hplc纯度>98%)在室温条件下溶于22ml2%的氨水溶液中,磷酸泰地唑胺与氨水溶液中氨水的摩尔比为1:2,得磷酸泰地唑胺铵盐粗品3.8g,得到的磷酸泰地唑胺铵盐容易过滤,不结块,易烘干。esi-ms448.89[m-h]-;1hnmr(dmso,400mhz)δ:8.92(s,1h),8.20(m,2h),7.73(m,2h),7.52(d,1h),7.26(brs,5h),4.90(m,1h),4.48(s,3h),4.17(t,1h),3.99(t,1h),3.94(m,2h)。实施例2将磷酸泰地唑胺粗品5g(hplc纯度>98%)在室温条件下溶于220ml0.1%的氨水溶液中,磷酸泰地唑胺与氨水溶液中氨水的摩尔比为1:1,得磷酸泰地唑胺铵盐粗品3.2g,得到的磷酸泰地唑胺铵盐容易过滤,不结块,易烘干。esi-ms448.89[m-h]-;1hnmr(dmso,400mhz)δ:8.92(s,1h),8.20(m,2h),7.73(m,2h),7.52(d,1h),7.26(brs,5h),4.90(m,1h),4.48(s,3h),4.17(t,1h),3.99(t,1h),3.94(m,2h)。实施例3将磷酸泰地唑胺粗品5g(hplc纯度>98%)在室温条件下溶于11ml18%的氨水溶液中,磷酸泰地唑胺与氨水溶液中氨水的摩尔比为1:9,得磷酸泰地唑胺铵盐粗品4.3g,得到的磷酸泰地唑胺铵盐容易过滤,不结块,易烘干。esi-ms448.89[m-h]-;1hnmr(dmso,400mhz)δ:8.92(s,1h),8.20(m,2h),7.73(m,2h),7.52(d,1h),7.26(brs,5h),4.90(m,1h),4.48(s,3h),4.17(t,1h),3.99(t,1h),3.94(m,2h)。实施例4分别取磷酸泰地唑胺粗品及实施例1中磷酸泰地唑胺铵盐分别充分研细,室温下分别用去离子水配制成饱和溶液(超声30min,溶液中仍有白色固体存在),用紫外分光光度法测定饱和溶液中溶质的含量,实验结果见表4。表4磷酸泰地唑胺磷酸泰地唑胺铵盐溶解度(g/l)0.42从表4可以看出,磷酸泰地唑胺铵盐在水中的溶解度是磷酸泰地唑胺粗品的5倍。实施例5将磷酸泰地唑胺粗品500g(hplc纯度>98%)在室温条件下溶于1.1l2%的氨水溶液中,磷酸泰地唑胺与氨水溶液中氨水的摩尔比为1:1,过滤;在30℃条件下,滴加3m甲酸调节ph至4,搅拌析晶,控制搅拌速度为170rpm/min,析晶时间为6h,过滤,60℃真空干燥,得400g白色晶体,hplc纯度为99.90%。该晶体的x-射线粉末衍射、红外、dsc、tga以及粒度分布谱图详见图1-5,在本发明中将其命名为磷酸泰地唑胺铵盐晶型a。实施例6将磷酸泰地唑胺粗品1g(hplc纯度>98%)在室温条件下溶于22ml1%的氨水溶液中,磷酸泰地唑胺与氨水溶液中氨水的摩尔比为1:5,过滤;在5℃条件下,滴加0.1m甲酸调节ph至3.5,搅拌析晶,控制搅拌速度为170rpm/min,析晶时间为6h,过滤,60℃真空干燥,得0.66g白色晶体,hplc纯度99.90%。经测x-射线粉末衍射图谱(xrd),确认为磷酸泰地唑胺铵盐晶型a。实施例7将磷酸泰地唑胺粗品1g(hplc纯度>98%)在室温条件下溶于11ml10%的氨水溶液中,磷酸泰地唑胺与氨水溶液中氨水的摩尔比为1:25,过滤;在20℃条件下,滴加1m甲酸调节ph至3.2,搅拌析晶,控制搅拌速度为170rpm/min,析晶时间为12h,过滤,60℃真空干燥,得0.75g白色晶体,hplc纯度99.89%。经测x-射线粉末衍射图谱(xrd),确认为磷酸泰地唑胺铵盐晶型a。实施例8将磷酸泰地唑胺粗品1g(hplc纯度>98%)在室温条件下溶于7ml5%的氨水溶液中,磷酸泰地唑胺与氨水溶液中氨水的摩尔比为1:8,过滤;在40℃条件下,滴加1m乙酸调节ph至3.0,搅拌析晶,控制搅拌速度为170rpm/min,析晶时间为12h,过滤,60℃真空干燥,得0.68g白色晶体,hplc纯度99.90%。经测x-射线粉末衍射图谱(xrd),确认为磷酸泰地唑胺铵盐晶型a。实施例9将磷酸泰地唑胺粗品1g(hplc纯度>98%)在室温条件下溶于15ml3%的氨水溶液中,磷酸泰地唑胺与氨水溶液中氨水的摩尔比为1:10,过滤;在30℃条件下,滴加1m乙酸调节ph至3.5,搅拌析晶,控制搅拌速度为170rpm/min,析晶时间为8h,过滤,60℃真空干燥,得0.52g白色晶体,hplc纯度99.89%。经测x-射线粉末衍射图谱(xrd),确认为磷酸泰地唑胺铵盐晶型a。实施例10将磷酸泰地唑胺粗品1g(hplc纯度>98%)在室温条件下溶于4ml25%的氨水溶液中,磷酸泰地唑胺与氨水溶液中氨水的摩尔比为1:23,在30℃条件下,滴加0.5m甲酸调节ph至3.5,搅拌析晶,控制搅拌速度为170rpm/min,析晶时间为6h,过滤,60℃真空干燥,得0.78g白色晶体,hplc纯度99.90%。经测x-射线粉末衍射图谱(xrd),确认为磷酸泰地唑胺铵盐晶型a。实施例11将磷酸泰地唑胺粗品1g(hplc纯度>98%)在室温条件下溶于90ml0.1%的氨水溶液中,磷酸泰地唑胺与氨水溶液中氨水的摩尔比为1:2,过滤;在20℃条件下,滴加0.5m甲酸调节ph至3.5,搅拌析晶,控制搅拌速度为170rpm/min,析晶时间为6h,过滤,60℃真空干燥,得0.88g白色晶体,hplc纯度99.90%。经测x-射线粉末衍射图谱(xrd),确认为磷酸泰地唑胺铵盐晶型a。实施例12将磷酸泰地唑胺粗品1g(hplc纯度>98%)在室温条件下溶于11ml8%的氨水溶液中,磷酸泰地唑胺与氨水溶液中氨水的摩尔比为1:20,过滤;在30℃条件下,滴加3m甲酸调节ph至3.2,搅拌析晶,控制搅拌速度为170rpm/min,析晶时间为6h,过滤,60℃真空干燥,得0.51g白色晶体,hplc纯度99.90%。经测x-射线粉末衍射图谱(xrd),确认为磷酸泰地唑胺铵盐晶型a。实施例13将磷酸泰地唑胺铵盐粗品1g(hplc纯度>98.5%)在室温条件下溶于500ml水中,过滤;在30℃条件下,滴加1m甲酸调节ph至3,搅拌析晶,控制搅拌速度为170rpm/min,析晶时间为6h,过滤,60℃真空干燥,得0.56g白色晶体,hplc纯度99.89%。经测x-射线粉末衍射图谱(xrd),确认为磷酸泰地唑胺铵盐晶型a。实施例14将磷酸泰地唑胺铵盐粗品1g(hplc纯度>98.5%)在室温条件下溶于600ml的水中,过滤;在5℃条件下,滴加0.1m甲酸调节ph至4,搅拌析晶,控制搅拌速度为170rpm/min,析晶时间为6h,过滤,60℃真空干燥,得0.36g白色晶体,hplc纯度99.91%。经测x-射线粉末衍射图谱(xrd),确认为磷酸泰地唑胺铵盐晶型a。实施例15将磷酸泰地唑胺铵盐粗品1g(hplc纯度>98%)在室温条件下溶于400ml水中,过滤;在40℃条件下,滴加3m乙酸调节ph至3.2,搅拌析晶,控制搅拌速度为170rpm/min,析晶时间为6h,过滤,60℃真空干燥,得0.48g白色晶体,hplc纯度99.90%。经测x-射线粉末衍射图谱(xrd),确认为磷酸泰地唑胺铵盐晶型a。实施例16将磷酸泰地唑胺铵盐粗品1g(hplc纯度>98.5%)在室温条件下溶于42ml0.1%的氨水溶液中,磷酸泰地唑胺铵盐与氨水溶液中氨水的摩尔比为1:1,过滤;在20℃条件下,滴加0.1m甲酸调节ph至3.4,搅拌析晶,控制搅拌速度为170rpm/min,析晶时间为6h,过滤,60℃真空干燥,得0.46g白色晶体,hplc纯度99.90%。经测x-射线粉末衍射图谱(xrd),确认为磷酸泰地唑胺铵盐晶型a。实施例17将磷酸泰地唑胺铵盐粗品1g(hplc纯度>98.5%)在室温条件下溶于4.2ml2%的氨水溶液中,磷酸泰地唑胺铵盐与氨水溶液中氨水的摩尔比为1:2,过滤;在40℃条件下,滴加1m甲酸调节ph至4,搅拌析晶,控制搅拌速度为170rpm/min,析晶时间为6h,过滤,60℃真空干燥,得0.35g白色晶体,hplc纯度99.91%。经测x-射线粉末衍射图谱(xrd),确认为磷酸泰地唑胺铵盐晶型a。实施例18将磷酸泰地唑胺铵盐粗品1g(hplc纯度>98%)在室温条件下溶于3.5ml18%的氨水溶液中,磷酸泰地唑胺铵盐与氨水溶液中氨水的摩尔比为1:15,过滤;在5℃条件下,滴加3m乙酸调节ph至3,搅拌析晶,控制搅拌速度为170rpm/min,析晶时间为6h,过滤,60℃真空干燥,得0.58g白色晶体,hplc纯度99.89%。经测x-射线粉末衍射图谱(xrd),确认为磷酸泰地唑胺铵盐晶型a。实施例6-18所得产物的x-射线粉末衍射谱图与实施例5相同,在此不再重复示出。制备例1:参照专利wo2015158202a1实施例1的制备方法得到磷酸泰地唑胺的晶型(命名为晶型i),具体操作步骤如下:取20g磷酸泰地唑胺,在80℃下用1ln,n-二甲基甲酰胺溶清,恒温保持10min,之后置于80℃下挥发至干,得到19.6g白色固体产物,产率98%。制备例2:参照专利cn102439006a实施例3的制备方法得到磷酸泰地唑胺的晶型(命名为晶型ii),具体操作步骤如下:将10g磷酸泰地唑胺二钠盐溶于50ml水中缓慢加入hcl(40ml;1m),得到淡黄色固体的良好的混悬液,再向其中加入25ml水。将固体过滤,用50ml0.1m的hcl和100ml水洗涤,真空干燥,得8.2g,产率82%。对比例1将制备例1中制备得到的晶型i、制备例2中制备得到的晶型ii和本发明实施例5中所得的晶型a,分别进行过滤速度对比实验、澄清度对比实验、75%rh条件下的吸湿性对比实验及室温条件下在水中的溶解度的对比实验。过滤速度对比实验:真空度为-0.09mpa,布氏漏斗为在过滤装置中流动的液体的过滤流量(ml/min)与过滤装置中的过滤器膜的过滤总面积(cm2)之比即为该过滤装置中的液体的过滤速度(ml/min/cm2)。澄清度对比实验:取1g磷酸泰地唑胺铵盐加入容量瓶中,加入碳酸钠溶液,使钠离子与磷酸泰地唑胺的摩尔比为2:1,将溶液定容至20ml,进行浊度测试。吸湿性对比试验:取干燥的具塞玻璃称量瓶(外径为50mm,高为15mm),于试验前一天置于适宜的25℃±1℃恒温干燥器(下部放置氯化钠饱和溶液),分别取同样重量的制备例1中制备得到的晶型i、制备例2中制备得到的晶型ii和本发明实施例5中所得的晶型a,平铺于上述称量瓶中,在温度为25℃±1℃、相对湿度为75%±2%条件下放置24h,进行引湿性实验。溶解度对比实验:分别取制备例1中制备得到的晶型i、制备例2中制备得到的晶型ii和本发明实施例5中所得的晶型a分别充分研细,室温下分别用去离子水配制成饱和溶液(超声30min,溶液中仍有白色固体存在),用紫外分光光度法测定饱和溶液中溶质的含量。实验结果见表5。表5从表5中的过滤速度、澄清度、吸湿性及溶解度的对比实验可以看出,本发明所得的晶型a的过滤速度、澄清度、吸湿性及溶解度均优于晶型ⅰ和晶型ⅱ。对比例2取制备例1中制备得到的晶型i、制备例2制备得到的晶型ii和本发明实施例5中所得的晶型a,进行室温光照条件下放置10天的稳定性实验。光照条件为4500lx照度,检测化合物在放置前后的hplc纯度和最大单杂含量,结果见表6。表6从表6的室温、4500lx照度光照条件下放置10天的稳定性实验显示:10天后,晶型a的hplc纯度变化值和最大单杂含量变化值均小于晶型ⅰ和晶型ⅱ,该结果表明,本发明获得的晶型a比晶型ⅰ和晶型ⅱ更稳定。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1