本发明属于药物合成技术领域,尤其涉及一种丁苯酞的制备方法。
背景技术:
丁苯酞[(±)-3-正丁基苯酞]是我国具有自主知识产权的国家一类新药。它和芹菜籽油中提取的左旋芹菜甲素[(-)-(s)-3-正丁基苯酞]的结构相同,为其人工合成的消旋体,具有广谱抗惊厥活性。临床研究结果表明,丁苯酞对急性和恢复期缺血性脑卒中患者有明显治疗效果;能够改善脑能量代谢,增加缺血区脑血流量,改善脑供血,缩小脑梗塞面积,保护和修复缺血区脑神经细胞。其结构式如下:
丁苯酞(3-butylphthalide)软胶囊是石药集团恩必普药业有限公司上市的拥有自主知识产权的、全新化学结构的国家一类抗脑缺血新药,商品名为“恩必普”,其可通过降低花生四烯酸含量,提高脑血管内皮no和pgi2的水平,抑制谷氨酸释放,降低细胞内钙浓度,抑制自由基和提高抗氧化酶活性等机制而对于脑缺血所致的多个病理环节产生药效作用。临床上可用于轻、中度急性缺血性脑卒中。
现有技术中,李绍白,张少明等在1990年兰州大学学报(自然科学版)上发表,以邻苯二甲酸酐、乙酸钠、戊酸酐在300℃加热回流制备丁烯苯酞粗品,采用硅胶柱层析进行精制,收率25%,收率明显偏低。以乙醚作为氢化溶剂,pd/c为催化剂,还原制得丁苯酞,进行柱层析得到丁苯酞该步收率95%。但该方法涉及高温和乙醚的使用,以及精制过程中均采用柱层析,不适合工业化生产。该方法工艺路线如下:
中国协和医科大学詹玉莲博士学位论文提到了采用邻苯二甲酸酐、戊酸酐、戊酸钠进行丁烯苯酞的合成,精馏后收率60~65%;以乙醇作为氢化溶剂,pd/c为催化剂制备丁苯酞粗品,蒸馏后收率90~95%,将得到的馏分在高回流比的减压蒸馏柱上进行精馏,精馏收率90~95%。该方法采用戊酸钠代替了乙酸钠,降低了反应温度,并避免了反应过程中乙酸蒸汽对环境的污染;催化采用乙醇替代乙醚,安全性大大提高,但其仍需要高精馏度多次蒸馏,能源消耗大,总成本较高,不利于生产。该方法工艺路线如下:
公开号为cn105884726a的中国专利公开了一种丁苯酞的合成方法和纯化工艺,其采用邻甲酰基苯甲酸为起始原料,以thf为溶剂和正丁基氯化镁格式试剂反应,调酸后制备得到丁苯酞产品;将丁苯酞粗品用碱性物质进行水解处理,再通过酸析出固体,过滤,得丁苯酞中间体,其主要杂质为未反应起始原料和苯酞;重复上述调酸调碱过程,最后进行关环、减压脱溶得到高纯度的丁苯酞,纯化收率为48~52%。该方法纯化工艺操作简单,不需要柱层析及高温、高真空减压精馏,易于工业化方法生产;但调酸过程中水中析出的中间板结不易从反应釜中放出,且丁苯酞中间体极不稳定,在固体情况下也会发生自身环合,多次调酸调碱会导致收率偏低,总成本较高。该方法工艺路线如下:
公开号为cn105130934a的中国专利公开了一种丁苯酞原料药产品及其制备方法,将丁苯酞粗品在甲醇中采用氢氧化钾进行水解,降温后加入醚类溶剂,使羟戊基苯甲酸钾盐析出;将该钾盐溶于水中,加入醚类溶剂搅拌析晶进行精制;随后将精制后的钾盐加入到水与二氯甲烷的混合液中,控制ph在1.5~3.0,温度30~45℃下酸化关环得到丁苯酞粗品,在精馏塔中,控制真空度4~5mmhg,升温至154~160℃,控制回流比3~7:1,得到丁苯酞原料药。该方法中羟戊基苯甲酸钾盐于水中的溶解度极高,其用醚类溶剂析晶收率低,且醚类溶剂成本较高,导致生产成本偏高。
公开号为cn105859670a的中国专利中公开了一种高纯度丁苯酞的制备方法,将丁苯酞粗品在真空度1-2mbar下减压,收集130~140℃馏分;将该馏分加入甲醇中采用无机碱水解,浓缩除去甲醇,加入二氯甲烷搅拌析出固体得羟戊基苯甲酸盐;随后将精制后的钾盐加入到水中,温度30~45℃下酸化关环得到丁苯酞,hplc纯度≥99.90%。但该方法仍需通过一次减压精馏,在经过成盐、二氯甲烷洗涤得到合格的丁苯酞,并不能避免减压精馏的过程。
技术实现要素:
有鉴于此,本发明要解决的技术问题在于提供一种丁苯酞的制备方法,该方法收率较高、纯度较高且无需精馏。
本发明提供了一种丁苯酞的制备方法,包括:
s1)以雷尼镍为催化剂,将丁烯基苯酞在醇溶剂中进行加氢反应,减压除去溶剂后,得到第一中间体;
s2)将所述第一中间体、氢氧化钠与水混合反应,然后用二氯甲烷洗涤反应体系,再加入醇溶剂,调酸反应后,得到第二中间体;
s3)将所述第二中间体在酸性的有机溶剂中进行合环反应,得到丁苯酞。
优选的,所述步骤s1)中加氢反应的温度为15℃~50℃;加氢反应的压力为1~10atm。
优选的,所述步骤s1)中丁烯基苯酞与醇溶剂的质量体积比为1g:(2~20)ml;所述步骤s1)中雷尼镍的质量为丁烯基苯酞质量的5%~20%。
优选的,所述第一中间体与水的质量体积比为1g:(2~20)ml;第一中间体与醇溶剂的质量体积比为1g:(0.1~1)ml;所述丁烯基苯酞与二氯甲烷的质量体积比为1g:(2~20)ml。
优选的,所述步骤s2)中混合反应的温度为10℃~80℃,混合反应的时间为1~3h。
优选的,所述步骤s2)中采用冰醋酸调酸;调酸至ph值为4~6;调酸反应的温度为0℃~20℃。
优选的,所述步骤s3)中合环反应的温度为20℃~40℃;所述步骤s3)中合环反应的ph值为1~4。
优选的,所述丁烯基苯酞按照以下方法制备:
将戊酸酐、邻苯二甲酸酐与部分戊酸钠混合反应,然后加入余下的戊酸钠继续反应;
反应结束后用氨水调节反应体系的ph值为碱性,再用二氯甲烷提取,得到丁烯基苯酞。
优选的,所述步骤s2)具体为:
将所述第一中间体、氢氧化钠与水混合在10℃~80℃反应,反应结束后使体系温度为10℃~30℃,然后用二氯甲烷洗涤反应体系,再加入醇溶剂,调酸反应后,得到第二中间体。
优选的,所述步骤s3)具体为:
将所述第二中间体在酸性的有机溶剂中进行合环反应,反应0.5~2h后,分出有机层,再补加有机溶剂,继续反应,最后合并有机层,得到有机相;
将所述有机相依次经弱碱性溶液洗涤、水洗、干燥剂干燥后,回收溶剂,得到丁苯酞。
本发明提供了一种丁苯酞的制备方法,包括:s1)以雷尼镍为催化剂,将丁烯基苯酞在醇溶剂中进行加氢反应,减压除去溶剂后,得到第一中间体;s2)将所述第一中间体、氢氧化钠与水混合反应,然后用二氯甲烷洗涤反应体系,再加入醇溶剂,调酸反应后,得到第二中间体;s3)将所述第二中间体在酸性的有机溶剂中进行合环反应,得到丁苯酞。与现有技术相比,本发明采用醇溶剂、二氯甲烷作为反应或萃取溶剂,其均可有效回收且价格便宜,大大降低了生产成本,再经过水解纯化、调酸过滤纯化,即可得较高收率的得到高纯度的符合制剂要求的丁苯酞,无需减压精馏步骤,操作简单。
附图说明
图1为本发明实施例4中得到的丁烯苯酞的hplc图谱;
图2为本发明实施例5中得到的第一中间体的hplc图谱;
图3为本发明实施例6中得到的丁苯酞的hplc图谱。
具体实施方式
下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明提供了一种丁苯酞的制备方法,包括:s1)以雷尼镍为催化剂,将丁烯基苯酞在醇溶剂中进行加氢反应,减压除去溶剂后,得到第一中间体;s2)将所述第一中间体、氢氧化钠与水混合反应,然后用二氯甲烷洗涤反应体系,再加入醇溶剂,调酸反应后,得到第二中间体;s3)将所述第二中间体在酸性的有机溶剂中进行合环反应,得到丁苯酞。
本发明对所有原料的来源并没有特殊的限制,为市售或自制即可。
在本发明中,所述丁烯基苯酞优选按照以下方法制备:将戊酸酐、邻苯二甲酸酐与部分戊酸钠混合反应,然后再加入余下的戊酸钠继续反应;反应结束后用氨水调节反应体系的ph值为碱性,再用二氯甲烷提取,得到丁烯基苯酞。
本发明以戊酸酐、邻苯二甲酸酐与戊酸钠为原料进行丁烯苯酞的合成,并且将戊酸钠分批加入,降低了在加热过程中反应骤然加剧释放出大量气体冲出反应釜的危险;其中,所述戊酸酐、邻苯二甲酸酐与戊酸钠的摩尔比优选为1:(0.8~1.2):(0.8~1.2),更优选为1:(0.9~1.1):(0.8~1),再优选为1:(1~1.1):(0.8~0.9),最优选为1:1.1:0.8。
将戊酸钠、邻苯二甲酸酐与部分戊酸钠混合反应,所述反应的温度优选为160℃~180℃,更优选为165℃~175℃,再优选为170℃;所述反应的时间优选为0.5~2h,更优选为1~1.5h,再优选为1h;然后加入余下的戊酸钠继续反应;所述部分戊酸钠与余下的戊酸钠的质量比优选为(1~2):(1~2),更优选为1:1;所述继续反应的温度优选为170℃~200℃,更优选为180℃~200℃,再优选为185℃~195℃,最优选为190℃;所述继续反应的时间优选为2~5h,更优选为2~4h,再优选为3h。
继续反应结束后,优选将反应体系的温度降至90℃~110℃,更优选降至95℃~105℃,再优选降至100℃,再加入水,继续加热进行回流反应。所述水加入的体积优选为戊酸酐质量的1~4倍,更优选为2~3倍,再优选为2倍;所述回流反应的时间优选为0.5~2h,更优选为1~1.5h,再优选为1h。
反应结束后,优选降至20℃~40℃,更优选降至20℃~35℃,再优选降温至25℃~30℃,最优选为25℃,用氨水调节反应体系的ph值为碱性,更优选为8~10,再优选为8.5~9.5,最优选为9;再用二氯甲烷提取;所述提取的次数优选为2~4次,更优选为3次。
提取后,优选依次经稀氨水洗涤、去离子水洗涤与干燥剂干燥后,回收二氯甲烷,得到丁烯基苯酞;所述稀氨水洗涤的次数优选为2~3次;所述去离子水洗涤的次数优选为2~3次;所述干燥剂为本领域技术人员熟知的干燥剂即可,并无特殊的限制,本发明中优选为无水氯化钙。
得到的丁烯基苯酞无需精馏,可直接进行加氢反应;还可进行减压蒸馏,收集140~145℃/2~3mmhg馏分,得到黄色油状液体,纯度98%左右,收率70%~75%。
以雷尼镍为催化剂,将丁烯基苯酞在醇溶剂中进行加氢反应,优选先将丁烯基苯酞与醇溶剂混合,再加入雷尼镍,进行加氢反应;所述醇溶剂为本领域技术人员熟知的醇溶剂即可,并无特殊的限制,本发明中优选为乙醇,更优选为无水乙醇;所述丁烯基苯酞与醇溶剂的质量体积比优选为1g:(2~20)ml,更优选为1g:(3~15)ml,再优选为1g:(3~10ml),再优选为1g:(3~8ml),最优选为1g:(4~6ml);所述雷尼镍的质量优选为丁烯基苯酞质量的5%~20%,更优选为10%~15%;所述加氢反应的温度优选为15℃~50℃,更优选为20℃~40℃,再优选为25℃~35℃;所述加氢反应的压力优选为1~10atm(1~10kg/cm2),更优选为1~8atm,再优选为1.5~6atm,再优选为1.5~5atm,再优选为1.5~3.5atm,最优选为2.5atm;至不吸氢,反应结束。
反应结束后,优选除去催化剂,再减压除去溶剂后,得到淡黄色油状液体为第一中间体,即丁苯酞粗品。
将所述第一中间体、氢氧化钠与水混合反应,优选先将氢氧化钠与水混合,然后在搅拌的条件下,滴加第一中间体;所述第一中间体与水的质量体积比优选为1g:(2~20)ml,更优选为1g:(3~15)ml,再优选为1g:(3~10ml),再优选为1g:(3~8ml),最优选为1g:(4~6ml);所述混合反应的温度优选为10℃~80℃,更优选为35℃~80℃,再优选为50℃~60℃;所述混合反应的时间优选为1~3h,更优选为2~3h,再优选为2h。
反应结束后,优选使反应体系的温度为10℃~30℃,更优选为20℃~30℃,再优选为20℃~25℃,然后用二氯甲烷洗涤反应体系,得到黄色清液;再加入醇溶剂,调酸进行反应;所述醇溶剂为本领域技术人员熟知的醇溶剂即可,并无特殊的限制,本发明中优选为乙醇,更优选为无水乙醇;所述第一中间体与醇溶剂的质量体积比优选为1g:(0.1~1)ml,更优选为1g:(0.2~0.8)ml,再优选为1g:(0.3~0.5)ml;所述调酸优选采用有机酸进行,更优选采用冰醋酸;所述调酸优选使反应体系的ph值为4~6,更优选为5~6,再优选为6;所述调酸反应的温度优选为0℃~20℃,更优选为0℃~15℃,再优选为0℃~10℃。调酸反应结束后,优选过滤,更优选进行甩滤,得到第二中间体,即羟戊基苯甲酸。
将所述第二中间体在酸性的有机溶剂中进行合环反应;其中,所述有机溶剂为本领域技术人员熟知的有机溶剂即可,并无特殊的限制,本发明中优选为二氯甲烷;所述酸性的有机溶剂优选使用无机酸提供酸性条件,更优选使用盐酸提供酸性条件;所述合环反应的温度优选为20℃~40℃,更优选为25℃~35℃;所述合环反应的ph值优选为1~4,更优选为1~3,再优选为1~2;在本发明中,该步骤优选具体为:将所述第二中间体在酸性的有机溶剂中进行合环反应,反应0.5~2h后,优选反应0.5~1h后,分出有机层,再补加有机溶剂,继续反应,最后合并有机层,得到有机相;将所述有机相依次经弱碱性溶液洗涤、水洗、干燥剂干燥后,回收溶剂,得到丁苯酞。所述继续反应的时间优选为0.5~2h,更优选为0.5~1h;所述弱碱性溶液为本领域技术人员熟知的弱碱性溶液即可,并无特殊的限制,本发明中优选为稀氨水;所述干燥剂为本领域技术人员熟知的干燥剂即可,并无特殊的限制,本发明中优选为无水氯化钙;所述回收溶剂的方法为本领域技术人员熟知的方法即可,并无特殊的限制,本发明中优选为减压浓缩回收溶剂。在此步骤中,第二中间体分批加入,由于反应始终处于酸性环境下,第二中间体直接关环,不会影响搅拌。
本发明以水作为水解反应溶剂,通过二氯甲烷萃取去除杂质,再经过乙醇调酸析出第二中间体,过滤纯化,也起到精制作用,不需进一步精制,酸化合环后即可得到符合制剂要求的丁苯酞原料药。
本发明提供的丁苯酞的制备工艺如下:
本发明采用醇溶剂、二氯甲烷作为反应或萃取溶剂,其均可有效回收且价格便宜,大大降低了生产成本,再经过水解纯化、调酸过滤纯化,即可得较高收率的得到高纯度的符合制剂要求的丁苯酞,无需减压精馏步骤,操作简单。并且,本发明经过小试、中试的工艺验证,能确保生产出来的产品质量符合原料药的要求,该工艺收率较高,具有良好的重现性和可行性。
为了进一步说本发明,以下结合实施例对本发明提供的一种丁苯酞的制备方法进行详细描述。
以下实施例中所用的试剂均为市售。
实施例1丁烯苯酞(dbt-1)的制备及蒸馏
将戊酸酐93.12g、邻苯二甲酸酐111.09g,戊酸钠24.82g,加入到1l反应瓶中,搅拌混合均匀,加热到170℃左右进行反应1h;随后补加戊酸钠24.82g,再升温到190℃左右,继续保温反应3h;反应毕,将体系降温至100℃左右,滴加186ml去离子水,继续加热至回流,继续回流1h,降温,降至室温;控制体系温度在25℃左右,采用浓氨水调ph至9,二氯甲烷450ml提取三次,采用150ml稀氨水(1%)洗涤两次,150ml去离子水洗涤两次,无水氯化钙干燥;抽滤,除去干燥剂,浓缩回收二氯甲烷,得深红色油状液体,20cm长刺型精馏柱进行减压蒸馏,收集140~145℃/2~3mmhg馏分,得到62.88g黄色油状液体,即为丁烯苯酞(dbt-1)。
利用高效液相色谱对实施例1中得到的丁烯苯酞(dbt-1)进行分析,其(z/e)纯度98.196%(hplc)左右,较大杂质由大到小排列为1.083%、0.449%、0.058%,该步骤收率74.8%。
实施例2羟戊基苯甲酸(dbt-2)的制备
向1l氢化釜中加入无水乙醇240ml、丁烯基苯酞(dbt-1)60.00g,加入雷尼镍(湿)9.00g,进行4次氢气交换排除空气。将温度加热到25~35℃,压力控制2.5个大气压(2.5kg/cm2)进行加氢反应,至不吸氢为止,约2h;除去催化剂,减压浓缩乙醇,得到淡黄色油状液体即为第一中间体,纯度96.324%。
向1l反应瓶中加入300ml去离子水、氢氧化钠13.20g,搅拌溶解,加入上述淡黄色油状液体,控温50~60℃,反应约2h;反应毕,降温到20℃左右用300ml二氯甲烷提取三次,水相中加入30ml无水乙醇,用冰醋酸调ph=6,抽滤,用30ml水洗涤滤饼得第二中间体(dbt-2)。
实施例3丁苯酞(dbt)的制备及精馏
向1l反应瓶中加入120ml2n盐酸、120ml二氯甲烷,调ph=1~2,控温25~35℃之间;将羟基戊基苯甲酸即第二中间体(dbt-2)分批加到反应体系中,控制体系温度25~35℃之间,反应30min。分出二氯甲烷层,继续补加120ml二氯甲烷,控制体系温度25~35℃之间,控制ph1~2之间,继续反应30min。合并有机层,采用稀氨水洗涤,去离子水洗涤,无水氯化钙干燥;减压浓缩回收溶剂,得26.43g浅黄色油状液体即为丁苯酞。
利用高效液相色谱对实施例3中得到的丁苯酞进行分析,得到其纯度99.718%,最大单杂0.070%。精制收率:43.6%。
实施例4丁烯苯酞(dbt-1)的制备及蒸馏
将戊酸酐441.41g、邻苯二甲酸酐386.59g,戊酸钠117.81g,加入到5l反应瓶中,搅拌混合均匀,加热到170℃左右进行反应1h;随后补加戊酸钠118.00g,再升温到190℃左右,继续保温反应3h;反应毕,将体系降温至100℃左右,滴加1l去离子水,继续加热至回流,继续回流1h,降温,降至室温;控制体系温度在25℃左右,采用浓氨水调ph至9,二氯甲烷2.5l提取三次,采用1l稀氨水(1%)洗涤两次,1l去离子水洗涤两次,100g无水氯化钙干燥;抽滤,除去干燥剂,浓缩回收二氯甲烷,得深红色油状液体,20cm长刺型精馏柱进行减压蒸馏,收集140~145℃/2~3mmhg馏分,得到339.66g黄色油状液体,即为丁烯苯酞(dbt-1)。
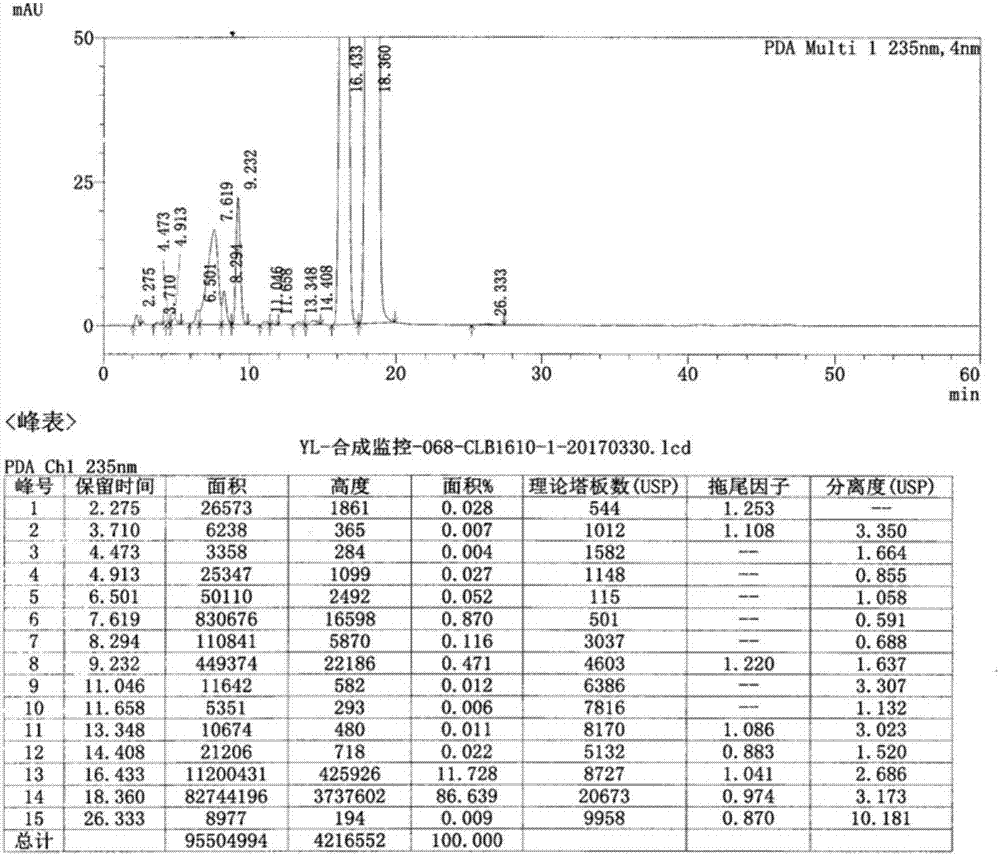
利用高效液相色谱对实施例1中得到的丁烯苯酞(dbt-1)进行分析,得到其hplc图谱,如图1所示。由图1可知,其(z/e)纯度98.367%(hplc)左右,较大杂质由大到小排列为0.870%、0.116%、0.471%,该步骤收率76.1%。
实施例5羟戊基苯甲酸(dbt-2)的制备
向氢化釜中加入无水乙醇1300ml、丁烯基苯酞(dbt-1)325.00g,加入雷尼镍(湿)48.75g,进行4次氢气交换排除空气。将温度加热到25~35℃,压力控制2.5个大气压(2.5kg/cm2)进行加氢反应,至不吸氢为止,约3h;除去催化剂,减压浓缩乙醇,得到淡黄色油状液体即为第一中间体,纯度97.893%。
向3l反应瓶中加入1600ml去离子水、氢氧化钠71.20g,搅拌溶解,加入上述淡黄色油状液体,控温50~60℃,反应约2h;反应毕,降温到20℃左右用1600ml二氯甲烷提取三次,水相中加入160ml无水乙醇,用冰醋酸调ph=6,抽滤,用160ml水洗涤滤饼得第二中间体(dbt-2)。
利用高效液相色谱对实施例2中得到的第一中间体进行分析,得到其hplc图谱,如图2所示。
实施例6丁苯酞(dbt)的制备及精馏
向2l反应瓶中加入650ml2n盐酸、650ml二氯甲烷,调ph=1~2,控温25~35℃之间;将羟基戊基苯甲酸即第二中间体(dbt-2)分批加到反应体系中,控制体系温度25~35℃之间,反应30min。分出二氯甲烷层,继续补加325ml二氯甲烷,控制体系温度25~35℃之间,控制ph1~2之间,继续反应30min。合并有机层,采用稀氨水洗涤,去离子水洗涤,无水氯化钙干燥;减压浓缩回收溶剂,得190.2g浅黄色油状液体即为丁苯酞。
利用高效液相色谱对实施例3中得到的丁苯酞进行分析,得到其hplc图谱,如图3所示。由图3可知,其纯度99.823%,最大单杂0.088%,总杂0.177%。精制收率:57.9%。