环丙沙星金属配合物及其制备方法和应用与流程
本发明涉及医药领域,特别涉及一种环丙沙星金属配合物及其制备方法和应用。
背景技术:
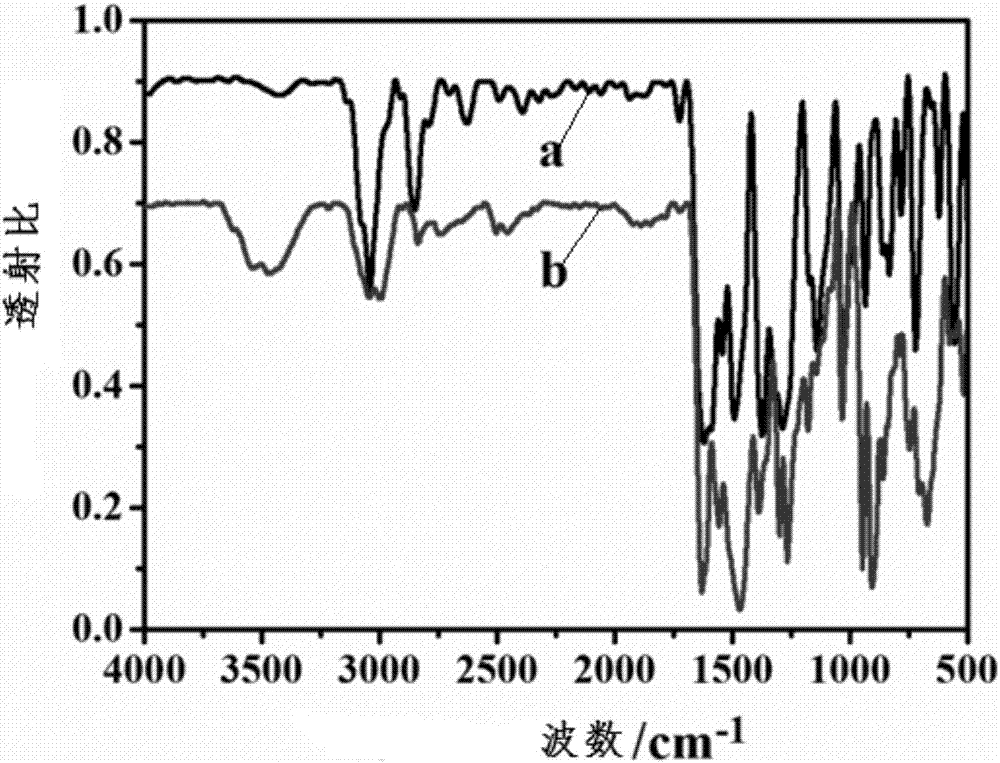
:喹诺酮类药物是一类人工合成的抗菌药。自从1962年美国研究人员首次发现喹诺酮类抗菌药萘啶酸以来,迄今为止已有近十万个喹诺酮类药物及其衍生物被合成出来,并进行了相关的生物活性研究。目前,喹诺酮类药物已经成为药物开发领域最具有价值的研究产品之一,已经被列入大多数家庭的常用药品清单中。但是随着喹诺酮类药物的使用日趋广泛,细菌耐药性趋势也随之日益增高,在此过程中会产生大量的耐药菌类,且耐药趋势不断加重。目前感染常见致病菌中革兰氏阳性菌对喹诺酮类药物的耐药性上升率较快,特别是耐甲氧西林金黄色葡萄球菌对环丙沙星的耐药率已高达到90%以上。因此需要从各方面着手,趋利避害,发挥其药效、利用其价值。。据相关文献报道,当具有生物活性的药物分子与金属离子形成配合物后,可以显著地改善药物的生物活性,进而降低细菌对药物的耐药性。虽然对环丙沙星配合物的生物活性研究报道日趋增多,但研究的对象几乎均是其简单配合物。而且对于抗菌剂的使用已经不再局限于在机体内部产生治疗效果;将其应用于器物表面,并且达到一个长效缓释的抗菌效果也是研究人员追求的目标。因此,制备新型的环丙沙星金属配合物,特别是筛选出抗菌性能明显优于环丙沙星本身的环丙沙星-金属配合物,并将其有效应用于生物体内和体外,发挥治疗效果具有重要意义。技术实现要素:为了解决上述问题,本发明人进行了锐意研究,结果发现:将环丙沙星、含有副族金属元素的化合物和多酸化合物混合,调节ph,升温反应后经后处理后得到环丙沙星金属配合物。该配合物有良好的生物活性,应用于多种日常器物表面抑菌、医疗器械表面抗菌和采用多种药物剂型进行抑菌等,尤其与聚烯醇进行复合,形成膜结构,其具有良好的缓释抗菌效果,从而完成了本发明。本发明的目的在于提供以下方面:第一方面,本发明提供一种环丙沙星金属配合物,其由环丙沙星与含有副族金属元素的化合物和多酸化合物配合而成,该配合物结构可如下表示:(z)z{x[mo(cf)n]m},其中z表示阳离子,x表示多酸阴离子,m表示副族金属元素。优选地,z为金属阳离子或铵根离子,x为含有过渡金属元素的多酸阴离子,优选为γ型多酸阴离子,更优选为含有vib族金属元素的多酸阴离子,尤其优选为含有铬元素、钼元素或钨元素的多酸阴离子中的一种,m为vb族金属元素,优选为钒元素、铌元素、钽元素中的一种,z为1~4,优选为1~3,例如2;n为1~4,优选为1~3,例如2;m为1~4,优选1~3,例如2。第二方面,本发明提供环丙沙星金属配合物的制备方法,该方法包括以下步骤:(1)将环丙沙星(cf)、含有副族金属元素的化合物和多酸化合物进行混合,任选搅拌;(2)向混合物或其溶液中加入ph调节剂,优选酸性物质,任选搅拌(3)升温进行反应;(4)反应结束后,进行后处理,得到目标产物。第三方面,根据第一方面的环丙沙星金属配合物的应用,尤其是用于抗菌,所述抗菌优选为日常器物表面抑菌、医疗器械表面抗菌和采用药物剂型进行抑菌。所述环丙沙星金属配合物与聚烯醇进行复合,优选形成膜结构,其结构优选为:(z)z{x[mo(cf)n]m}-p,p代表聚烯醇,优选为聚乙烯醇(pva)。所述环丙沙星金属配合物与聚烯醇复合物按如下方法进行制备:(1)按照第二方面制备环丙沙星金属配合物,及其溶液;(2)制备聚烯醇溶液;(3)制备环丙沙星金属配合物与聚烯醇复合物。根据本发明提供的环丙沙星金属配合物,具有以下有益效果:(1)环丙沙星与金属形成配合物后,可以通过协同作用增强环丙沙星生物活性,进而降低细菌对药物的耐药性。(2)所述环丙沙星金属配合物的物质结构丰富,其可应用于多种日常器物表面抑菌、医疗器械表面抗菌和采用多种药物剂型进行抑菌等,应用范围广泛。(3)将环丙沙星金属配合物负载到高分子材料上,尤其是聚烯醇材料上,作为一种高分子复合抗菌材料,可达到长效缓释的抗菌效果,使得释药速度稳定、药效提高、用药次数减少等;(4)本发明的制备方法简单,原料来源广泛,易于操作的同时,降低了生产成本,有利于产业化的推广。附图说明图1示出原料环丙沙星和实施例1的红外光谱图;图2a示出实施例2的光电子能谱及其v2p峰;图2b示出实施例2的光电子能谱及其mo3d峰;图3示出在扫描范围200~400nm的实施例2、对比例1、对比例2和对比例3的紫外吸收光谱图;图4示出实施例2、对比例1、对比例2在275nm激发波长下的紫外吸收光谱图;图5a示出实施例2(s)和对比例2(d)对大肠杆菌的缓释抗菌效果示意图;图5b示出实施例2(s)和对比例2(d)对金黄色葡萄球菌的缓释抗菌效果示意图。具体实施方式下面通过对本发明进行详细说明,本发明的特点和优点将随着这些说明而变得更为清楚、明确。在这里专用的词“示例性”意为“用作例子、实施例或说明性”。这里作为“示例性”所说明的任何实施例不必解释为优于或好于其它实施例。尽管在附图中示出了实施例的各种方面,但是除非特别指出,不必按比例绘制附图。根据本发明的第一方面,提供一种环丙沙星金属配合物,其由环丙沙星与含有副族金属元素的化合物和多酸化合物配合而成,所述环丙沙星金属配合物结构可如下表示:(z)z{x[mo(cf)n]m},其中,z表示阳离子,x表示多酸阴离子,m表示副族金属元素;在一个优选的实施方式中,z为金属阳离子或铵根离子,更优选为铵根离子(nh4+)。在一个优选的实施方式中,所述多酸阴离子为含有过渡金属元素的多酸阴离子,优选为γ型多酸阴离子,更优选为含有vib族金属元素的多酸阴离子,尤其优选为含有铬元素、钼元素或钨元素的多酸阴离子中的一种,进一步的优选为含有钼元素的多酸阴离子,如[γ-mo8o26]4-。在一个优选的实施方式中,m为vb族金属元素,优选为钒元素、铌元素、钽元素中的一种,更优选为钒元素。在一个优选的实施方式中,m为钒元素,其具有抗炎、杀菌等生物活性,钒化合物的生理效应及毒性与钒的总量、化合特性和形态有关,在合理的剂量范围内均对动物体的生理机能起促进作用,如维持生物体的生长、促进葡萄糖的吸收以及呈现出类胰岛素的作用等,当其与药物分子形成的配合物之后也具有较好的生物活性,可用于诊断和治疗某些疾病。钒在调节机体血糖代谢、泌尿代谢和心血管代谢等方面也发挥着重要的作用。在一个优选的实施方式中,x为含有钼元素的多酸阴离子,钼元素具有抗肿瘤活性、毒性低及多价态变化,使得配合物的结构更为丰富。在一个优选的实施方式中,z为1~4,优选为1~3,如2;n为1~4,优选为1~3,如2;m为1~4,优选1~3,如2。在一个优选的实施方式中,所述环丙沙星金属配合物以环丙沙星为有机配体,通过螯合配位的方式和副族金属元素进行配位。在一个优选的实施方式中,环丙沙星分子的羧基和酮羰基与副族金属元素进行配位,其中,羧基以单齿方式与金属元素进行配位。在一个优选的实施方式中,所述环丙沙星金属配合物的结构是以多酸阴离子为中心,通过端氧与环丙沙星和副族金属元素的配合物相连。根据本发明的第二方面,提供上述环丙沙星金属配合物的制备方法,其包括以下步骤:步骤(1),将环丙沙星(cf)、含有副族金属元素的化合物和多酸化合物进行混合,任选搅拌;在本发明中,所述含有副族金属元素的化合物优选为含有第vb族金属元素的化合物,更优选为含有钒元素、铌元素或钽元素的化合物中的一种,尤其优选为含有钒元素的化合物,如五氧化二钒。在一个优选的实施方式中,所述含有副族金属元素的化合物为含有副族金属元素的氧化物,优选为含有第vb族金属元素的氧化物,更优选为含有钒元素、铌元素或钽元素的氧化物中的一种,尤其优选为含有钒元素的氧化物,如五氧化二钒。在一个优选的实施方式中,所述含有副族金属元素的化合物可以通过购买或实验制得,优选实验自制,产物的纯度更高,有利于环丙沙星金属配合物的生成。在一个优选的实施方式中,所述含有副族金属元素的化合物为含有副族金属元素的氧化物,其由相应的含有副族金属元素的含氧酸盐制得,将含有副族金属元素的含氧酸盐加入到任选的可加热容器中,进行加热,任选搅拌,反应结束后冷却得到含有副族金属元素的氧化物,备用,其中,所述可加热容器优选为蒸发皿,加热方式优选为沙浴加热、水浴加热、加热套加热和电磁炉加热等,更优选沙浴加热;所述含有副族金属元素的含氧酸盐为含有第vb族金属元素的含氧酸盐,优选含有钒元素、铌元素或钽元素的含氧酸盐中的一种,更优选为含有钒元素的含氧酸盐,如偏钒酸铵。在一个优选的实施方式中,所述多酸化合物为多酸阴离子中含有过渡金属元素的化合物,优选为多酸阴离子中含有vib族金属元素的化合物,更优选为多酸阴离子中含有铬元素、钼元素或钨元素的化合物中的一种,进一步优选为多酸阴离子中含有钼元素的化合物,如(nh4)6mo7o24·4h2o。在一个优选的实施方式中,所述多酸化合物为模板剂,进而能形成以多酸阴离子为中心的配合物结构。优选地,所述含有副族金属元素的化合物与环丙沙星和多酸化合物的摩尔比为1:0.5~0.9:0.2~0.45,如1:0.7:0.35。在一个优选的实施方式中,在步骤(1)中还加入盐类强电解质溶液,优选为kcl溶液或nacl溶液等,更优选为kcl溶液。优选地,盐类强电解质的浓度为1~4mol·l-1,更优选1.5~3mol·l-1,如3mol·l-1。在本发明中,加入盐类强电解质以使得溶液中各种电解质平衡,保持相对稳定。优选地,环丙沙星与盐类强电解质溶液的重量体积比为(0.12~0.30)重量份:(1.6~4)体积份,更优选为(0.15~0.25)重量份:(2~3.2)体积份,如0.23重量份:3体积份,其中,基于1g计为1重量份,基于1ml计为1体积份。在一个优选的实施方式中,在步骤(1)中还加入溶剂,所述溶剂优选为水、有机溶剂或二者的组合物,更优选为水和有机溶剂的组合物,其中,所述有机溶剂优选为甲醇、乙醇、异丙醇、丙酮等,优选为乙醇。在本发明中,所述溶剂优选为有机溶剂和水的组合物,有机溶剂和水的体积比为1:1~4,优选为1:1.5~3,如1:2。优选地,环丙沙星与所加溶剂的重量体积比为(0.12~0.30)重量份:(10~30)体积份,更优选为(0.15~0.25)重量份:(12~20)体积份,如0.23重量份:15体积份,其中,基于1g计为1重量份,基于1ml计为1体积份。在一个优选的实施方式中,步骤(1)中的混合物或其溶液在15℃~35℃下反应搅拌1~4h,优选控制温度为20~30℃搅拌1.5~3h,进一步地,在25℃下搅拌2h。步骤(2),向混合物或其溶液中加入ph调节剂,优选酸性物质,调节ph为酸性,任选搅拌;在一个优选的实施方式中,所述ph调节剂为弱酸,优选为甲酸、乙酸、苯甲酸、乙二酸等,更优选为乙酸。在一个优选的实施方式中,ph调节剂的浓度为1~4mol·l-1,优选为1.5~3mol·l-1,如2mol·l-1。在一个优选的实施方式中,将混合物或其溶液的ph调节至3.5~5.5,优选地,调节ph至4,发明人发现,在ph<3时,环丙沙星的哌嗪基和羧基均被质子化,在ph>10时,哌嗪基和羧基均被去质子化,在中性和弱碱性情况下,哌嗪基的n原子能参与到金属的配位中去,但在弱酸条件下,哌嗪基的n原子不能参与到金属的配位中去,因而本发明优选ph调节至3.5~5.5。在一个优选的实施方式中,将步骤(2)中的溶液在15℃~30℃下搅拌10min~60min,优选地,控制温度为20~25℃搅拌1~1.5h,进一步地,在25℃下搅拌30min。步骤(3),升温进行反应;在一个优选的实施方式中,将步骤(3)得到的溶液转移至反应容器中,加热条件下恒温进行反应,所述反应容器优选为聚四氟乙烯低压反应釜。在一个优选的实施方式中,加热温度为100℃~150℃,优选为110~130℃,更优选为120℃。在一个优选的实施方式中,反应时间为2~5天,优选为4天。步骤(4),反应结束后,进行后处理,得到目标产物。在一个优选的实施方式中,反应结束后降温,可以采用自然降温或程序性降温,优选采用程序性降温,进一步地,以5~20k·h-1的速度程序性降温,优选的,降温速度为8~15k·h-1,更优选为10k·h-1。在本发明步骤(4)中,优选将反应结束后的物质降温至10℃~50℃,更优选为15℃~40℃,进一步优选为20℃~35℃,如25℃。在一个优选的实施方式中,将上述降温后得到的产物洗涤,除去其表面可能附着的可溶性杂质,用于洗涤固体的洗涤液优选为蒸馏水。在一个优选的实施方式中,在对降温后的产物进行洗涤后,对其进行干燥,优选使用真空干燥法、常压加热法、自然干燥法等,更优选使用真空干燥法进行干燥,由此得到本发明的目标产物,即环丙沙星金属配合物,其结构可如下表示:(z)z{x[mo(cf)n]m},其中,z表示阳离子,优选为金属阳离子或铵根离子,更优选为铵根离子;x表示多酸阴离子,优选为含有过渡金属元素的多酸阴离子,更优选为γ型多酸阴离子,尤其优选为含有vib族金属元素的多酸阴离子,进一步优选为含有铬元素、钼元素或钨元素的多酸阴离子中的一种,最优选为含有含有钼元素的的多酸阴离子,如(γ-mo8o26)4-;m表示副族金属元素,优选m为vb族金属元素,更优选为钒元素、铌元素、钽元素中的一种,尤其优选为钒元素;z为1~4,优选为1~3,如2;n为1~4,优选为1~3,如2;m为1~4,优选1~3,如2。红外光谱分析表明,该配合物与配体环丙沙星的吸收峰有所不同,cf在1724cm-1处出现的羧基特征伸缩振动吸收峰(νc=o),在形成配合物后,该处吸收峰消失,在1555cm-1、1388cm-1出现吸收峰,其对应于羧基的不对称伸缩振动和对称伸缩振动,说明cf中喹诺酮环上的羧基以单齿方式与钒配位,cf在1622cm-1处出现的羰基伸缩振动吸收峰(c=o),在形成配合物后,该处吸收峰红移至1629cm-1处,说明羰基也参与了配位。因此,本发明的环丙沙星金属配合物在1555cm-1、1388cm-1、1629cm-1处存在吸收峰。在本发明中,环丙沙星和金属离子之间存在协同作用,致配合物的抗菌活性强于环丙沙星,因此显著地改善药物的生物活性,进而降低细菌对药物的耐药性。根据本发明的第三方面,提供上述环丙沙星金属配合物的应用,尤其是用于抗菌。所述抗菌优选体外抗菌,更优选用于多种日常器物表面抑菌、医疗器械表面抗菌和采用多种药物剂型进行抑菌等。在应用中,所述环丙沙星金属配合物还可以与聚烯醇进行复合,优选形成膜结构。其结构优选为:(z)z{x[mo(cf)n]m}-p,其中,p代表聚烯醇,优选为聚乙烯醇(pva);以上复合物可按照以下方法进行制备:(1)按照上述第二方面制备环丙沙星金属配合物,及其溶液;在一种优选的实施方式中,将制得的环丙沙星金属配合物溶于溶剂中混合均匀,所述溶剂优选为蒸馏水;优选地,采用机械搅拌方式或是超声波振荡的方式使上述溶液充分混合均匀,更优选采用超声波振荡的方式,进一步地,混合时间优选为1~5h,更优选2~4h,如3h,使环丙沙星金属配合物均匀的分散在溶剂中;在一种优选的实施方式中,上述环丙沙星金属配合物与溶剂的重量体积比为(0.0080~0.020)重量份:(2~6)体积份,更优选为(0.0100~0.0015)重量份:(2~4)体积份,如0.0134重量份:3体积份,其中,基于1g计为1重量份,基于1ml计为1体积份。(2)制备聚烯醇溶液将聚烯醇溶于溶剂中加热,搅拌;在一种优选的实施方式中,将聚烯醇溶于蒸馏水中,加热搅拌至聚烯醇全部溶化,此时溶液呈胶状透明状态。优选地,加热温度为70~120℃,更优选为80~100℃,如90℃。在一种优选的实施方式中,上述聚烯醇与溶剂的重量体积比为(1~5)重量份:(10~50)体积份,更优选为(1.5~3)重量份:(15~30)体积份,如2重量份:20体积份,其中,基于1g计为1重量份,基于1ml计为1体积份。在一种优选的实施方式中,所述聚烯醇优选为聚乙烯醇,其是一种安全的生物可降解高分子有机物,对人体无毒,无副作用,具有良好的生物相容性,粘接强度好,其易成膜,且膜的机械性能优良,拉伸强度随聚合度、醇解度升高而增强。在一种优选的实施方式中,采用机械搅拌方式或是超声波振荡的方式处理上述聚烯醇溶液,优选采用超声震荡的方式,以除去溶液中的气泡。(3)制备复合物(z)z{x[mo(cf)n]m}-p将上述环丙沙星金属配合物配制成的溶液与聚烯醇溶液混合;在一个优选的实施方式中,将步骤(2)制备的聚烯醇溶液加入到步骤(1)制得的环丙沙星金属配合物溶液中,搅拌,优选剧烈搅拌1~4h,更优选1.5~3h,如3h,以便使二者均匀混合;在一个优选的实施方式中,步骤(3)中量取的环丙沙星金属配合物溶液与聚烯醇溶液的体积比为1:(0.5~2),更优选为1:(0.8~1.5),如1:1;在一种优选的实施方式中,采用机械搅拌方式或是超声波振荡的方式处理上述环丙沙星金属配合物溶液与聚烯醇溶液的混合液,优选采用超声震荡的方式,以除去混合液中的气泡。将混合液进行成膜;在一种优选的实施方式中,将上述制得的混合液干燥成膜,优选将混合液倒入成膜容器中后进行干燥成膜,进一步地,成膜容器优选为玻璃容器和塑料容器等,更优选为塑料容器,如96圆形孔板盖。在一个优选的实施方式中,将上述制得的混合液干燥成膜,优选使用真空干燥法、常压加热法、自然干燥法等,更优选使用真空干燥法。在一种优选的实施方式中,在40℃~80℃下进行干燥,更优选在45℃~60℃进行干燥,如50℃,进一步地,干燥时间优选为1~4h,更优选为1.5~3h,如2h。在一种优选的实施方式中,将干燥成型的复合膜剥离,收集,备用,该复合膜可表示为(z)z{x[mo(cf)n]m}-p,具体如上所述。所述复合膜具有缓释作用,其可以降低环丙沙星金属配合物的释放速率,使得释药速度稳定、药效提高、用药次数减少。在本发明中,发明人认为由于(z)z{x[mo(cf)n]m}中环丙沙星的哌嗪基上的氮可以与pva表面的羟基形成氢键,且多酸阴离子上的氧原子也可以与聚烯醇表面的羟基形成氢键,进而可以降低(z)z{x[mo(cf)n]m}从聚烯醇上的释放速率,从而达到缓释作用。实施例实施例1将0.085mol偏钒酸铵固体加入到蒸发皿中,置于沙浴上加热,不断搅拌,待白色固体全部变成红棕色固体停止加热,得到产物五氧化二钒(v2o5),冷却,待用。将1.00mmol上述制得的五氧化二钒、0.70mmol环丙沙星(cf)和0.35mmol(nh4)6mo7o24·4h2o混合,加入3mlkcl(3mol·l-1)溶液,再加入10mlh2o和5ml乙醇,在室温下搅拌2h。用2mol·l-1hac溶液将上述溶液的ph调至4.0,继续在室温下搅拌0.5h将搅拌均匀的浑浊液转移到25ml的聚四氟乙烯低压反应釜中,在120℃条件下恒温反应4天。以10k·h-1的速度程序性降温至室温。得到绿色块状晶体。蒸馏水洗涤,放置真空干燥箱中干燥,得到产物(nh4)2{(γ-mo8o26)[vo(cf)2]2}配合物,产率为35%(以v计),其x-射线单晶衍射如实验例1所述。使用元素分析仪测试(nh4)2{(γ-mo8o26)[vo(cf)2]2}配合物的结构中c、h和n的元素,实测结果为:c30.61、n7.36、h2.81与理论计算值c31.02、n7.33、h2.86,较好地吻合。测定原料环丙沙星和本实施例制得的(nh4)2{(γ-mo8o26)[vo(cf)2]2}配合物红外光谱图,测定范围4000~500cm-1,结果如图1所示,其中,曲线a示出原料环丙沙星制得样品的红外光谱图;曲线b示出实施例1制得样品的红外光谱图。由图1可知:环丙沙星在1724cm-1处出现的羧基特征伸缩振动吸收峰(νc=o)在实施例1中消失;而实施例1的羧基振动峰分别位于1555cm-1和1388cm-1。两者相差167cm-1,因为差值小于200cm-1,说明cf中喹诺酮环上的羧基以单齿方式与钒配位;原料cf在1622cm-1处出现的3位羰基伸缩振动吸收峰(c=o),在实施例1中红移至1629cm-1处,说明羰基也参与了配位;此外,实施例1位于946cm-1、860cm-1、789cm-1的特征吸收峰可归属为γ-mo8o26结构中ν(mo=o)和ν(mo-o-mo)的振动吸收峰;实施例1在3400cm-1附近还显示了h2o的强伸缩振动吸收峰;上述红外光谱的特征吸收峰的存在及变化趋势说明结构中有cf的金属配合物以及多酸化合物的存在。实施例2取0.0134g实施例1中制得的(nh4)2{(γ-mo8o26)[vo(cf)2]2},加入3ml蒸馏水,超声振荡3h,分散均匀,形成环丙沙星金属配合物溶液。将2.0g聚乙烯醇(pva)中加入20.0ml蒸馏水,在90℃加热搅拌至固体pva全部溶化,溶液呈胶状透明状态,然后采用超声波振荡法,将所有气泡除去,从而形成聚乙烯醇溶液。此时制得的pva溶液浓度为10%。在以上制得的环丙沙星金属配合物溶液中加入3ml以上制得的聚乙烯醇溶液,剧烈搅拌2h,然后进行超声振荡,将混合液中所有气泡除去。以96孔板盖的圆形凹槽为模板,取混合液30μl注入凹槽内,50℃真空干燥2h取出,将干燥成型的复合膜((nh4)2{(γ-mo8o26)[vo(cf)2]2}-pva),然后从孔板上剥离,收集,备用。对比例对比例1称取2.0g聚乙烯醇(pva)加入20.0ml蒸馏水,90℃加热搅拌至固体pva全部溶化,溶液呈胶状透明状态,然后采用超声波振荡法,将所有气泡除去,从而形成聚乙烯醇溶液。此时制得的pva溶液浓度为10%。量取3ml蒸馏水,再加入3ml上述制得的聚乙烯醇溶液,剧烈搅拌2h,聚乙烯醇分散均匀后,然后进行超声振荡,将混合液中所有气泡除去。以96孔板盖的圆形凹槽为模板,取混合液30μl注入凹槽内,50℃真空干燥2h取出,将干燥成型的聚乙烯醇膜,然后从孔板上剥离,收集,备用。对比例2称取0.0066g环丙沙星,再加入3ml蒸馏水,超声振荡3h,使环丙沙星均匀分散在水溶液中,形成环丙沙星溶液;称取2.0g聚乙烯醇(pva)加入20.0ml蒸馏水,90℃加热搅拌至固体pva全部溶化,溶液呈胶状透明状态,然后采用超声波振荡法,将所有气泡除去,从而形成聚乙烯醇溶液。此时制得的聚乙烯醇溶液浓度为10%。在上述制得的环丙沙星溶液中加入3ml上述制得的聚乙烯醇溶液,剧烈搅拌2h,确保环丙沙星水溶液和pva能够均匀混合,然后进行超声振荡,将混合液中所有气泡除去。以96孔板盖的圆形凹槽为模板,取混合液30μl注入凹槽内,50℃真空干燥2h取出,将干燥成型的复合膜(cf-pva),然后从孔板上剥离,收集,备用。对比例3方法与对比例2相同,仅区别为所用原料不是环丙沙星,而是(nh4)6mo7o24·4h2o,从而制得(nh4)6mo7o24·4h2o溶液,进行后续反应,最终制得[mo8o26]4--pva复合膜。实验例实验例1样品的x-射线单晶衍射本实验例所用样品为实施例1制得的样品。样品的x-射线单晶衍射数据是在带有多层膜的安捷伦supernova型ccdx-射线单晶衍射仪上用mokα射线()作为入射辐射,在293k温度下收集衍射数据。收集的数据经lp因子和经验吸收校正,结构采用直接法使用shelxtl软件包解析,由全矩阵最小二乘法优化,所有非氢原子坐标采用各向异性热参数修正。有机基团上的氢原子坐标采用几何加氢的方法得到,其中,样品的晶体学数据如表1所示:表1ar1=σ||fo|-|fc||/σ|fo|,bwr2=[σ[w(fo2-fc2)2]/σw(fo2)2]样品的键长和键角(°)如表2所示:表2由表1和表2分析可知,实施例1由两个单核钒配合物(简称v-cf2),一个[γ-mo8o26]4-(简称γ-mo8)多酸阴离子和两个nh4+阳离子组成。在单核钒配合物v-cf2结构中,v分别与2个环丙沙星4位羰基氧和3位羧酸上的羟基氧通过螯合配位的方式形成一个扭曲的{vo6}八面体,其中v=o键的键长为而其它v-o键的键长的距离位于范围内。多阴离子γ-mo8包括六个{moo6}八面体和两个{moo5}四方锥,其中在{moo6}八面体结构中mo-o键的距离位于之间。两组{moo6}八面体分别与两个{moo5}四方锥通过共边相连,在形成了交互的六元环。在γ-mo8单元中包含14个端氧(ot),6个双桥氧(μ2-o),4个三桥氧(μ3-o)和2个四桥氧(μ4-o)。mo-ot的平均键长在之间。mo-ob的平均键长在之间。γ-mo8分别与两个钒原子通过两个μ2-o相连构成了实施例1的单元结构,其中mo-o-v的键角为147.3(5)°。配合物的整体结构为以γ-[mo8o26]4-为中心,两端通过对称位置上的端氧分别与两个钒-环丙沙星配合物上的钒原子连接形成的夹心型结构。每个实施例1的单元分子通过γ-mo8上的端氧通过和环丙沙星分子哌嗪环上的氮原子形成的氢键(o1-h···n3,o17-h···n6)在空间沿b轴形成了1d有机超分子链,另一方面1d链通过cf配体之间的π···π堆积作用,两个芳香环中心距离为在空间形成了2d分子网络结构。实验例2样品的光电子能谱测定本实验例所用样品为实施例2制得的样品。采用多功能光电子能谱仪测定样品的光电子能谱。图2a示出实施例2的光电子能谱及其v2p峰;图2b示出实施例2的光电子能谱及其mo3d峰;由图2a所示,出现在516.15ev的峰归属于+5价态v的2p电子的特征峰,表明实施例2中v5+离子的存在。由图2b所示,,出现在232.6和235.6ev的两个峰归属于+6价态mo的特征峰,表明实施例2中mo6+离子的存在。实验例3样品抗菌性能测定本实验例所用样品为实施例1和原料环丙沙星制得的样品。样品的抗菌性能实验采取纸片法,实验方法参照国家标准实验方法,分别测定对大肠杆菌和金黄色葡萄球菌抑菌率,结果如表3和表4所示,表3实施例1和原料cf对大肠杆菌抑菌率样品第1次第2次第3次平均菌落抑菌率空白132128135132----原料cf998993.1实施例11311101191.7表4实施例1和原料cf对金黄色葡萄球菌抑菌率样品第1次第2次第3次平均菌落抑菌率空白135128131131----原料cf910101092.3实施例11211121290.8抑菌率=(空白组菌落数-实验组菌落数)/(空白菌落数)。可知,实施例1与原料环丙沙星的抑菌活性相近,但实施例1制得的配合物生物活性更好,且物质结构更丰富。实验例4样品的紫外可见光谱测定本实验例所用样品为实施例2、对比例1、对比例2和对比例3制得的样品。采用uv2550紫外可见分光光度仪,以石英为基片测定复合薄膜的紫外光谱,扫描范围200~400nm,结果如图3所示,其中,曲线a示出实施例2制得样品的紫外吸收光谱图;曲线b示出对比例1制得样品的紫外吸收光谱图;曲线c示出对比例2制得样品的紫外吸收光谱图;曲线d示出对比例3制得样品的紫外吸收光谱图。由图3可知,对比例2(cf-pva复合膜)的主要吸收带为272nm、320nm和332nm,对于实施例2((nh4)2{(γ-mo8o26)[vo(cf)2]2}-pva复合膜)而言,无论是最大吸收峰的位置、强度,还是峰的形状,都与cf-pva复合膜十分相似,显然该吸收带都应指配为cf的π→π*跃迁;与对比例2相比,实施例2最大吸收发生了微小紫移,不受任何理论束缚,本发明人认为,这是由于配体cf为大环分子,与钒离子配位后使分子不处于同一个平面上,分子的平面度降低,共轭性减少;同时钒离子对大π键又有一定吸引作用,配位后,环上电子云向钒离子移动,从而使得环上电荷分布不均匀,对称性降低,环的共轭性相应减小,波长发生紫移;此外,实施例2在200nm附近出现了新的特征吸收峰,该特征峰与对比例3在该处的特征吸收较吻合。综上,原料环丙沙星及(nh4)2{(γ-mo8o26)[vo(cf)2]2}成功的负载到pva薄膜上。实验例5样品体外缓释释放行为测定本实验例所用样品为实施例2、对比例1和对比例2制得的样品。复合膜的缓释效果通过测量每个时间段内化合物的累积释放量来进行监测。操作方法:分别取由实施例2、对比例1和对比例2制得的样品10片置于20ml烧杯中,加入10ml蒸馏水,37℃浸泡3h后将膜片取出,重新放置在新的20ml烧杯中,加入10ml蒸馏水,37℃条件下继续浸泡3h后再将膜片取出,在前12h内每3h进行上述操作一次,12h~48h内每12h重复取样一次。取样溶液收集在275nm激发波长下进行紫外吸光度测试,结果如图4所示,其中,曲线a示出实施例2制得样品的紫外吸收光谱图;曲线b示出对比例1制得样品的紫外吸收光谱图;曲线c示出对比例2制得样品的紫外吸收光谱图。由图4可知,在0h~9h内,对比例2的累计释放量达到了90%,9h之后,cf在水溶液中的释放量几乎没有变化,这表明cf在前9h之内有一个突释的过程,在此过程中,cf的累计释放量达到了最大值,9h之后,cf的释放量接近于零;实施例2的突释过程在0h~12h内,该过程中最大的释放量约27%,累计释放量达70%。12h~48h之间有一个缓慢释放的过程,释放累积量保持在9%~11%之间,且实施例2在48h后仍保持活性,抑菌率维持在35%。与对比例2在该过程中(0h-48h)的释放情况相比,实施例2在水溶液中有一个更缓慢的释放过程。综上,实施例2((nh4)2{(γ-mo8o26)[vo(cf)2]2}-pva复合膜)在水溶液中出现的缓释现象,不受任何理论束缚,发明人认为,因为实施例2结构中含有多酸基团[γ-mo8o26]4-,多酸表面含有大量的氧原子,这使得γ-mo8具有一个富氧结构,除了环丙沙星的哌嗪基上的氮以外,γ-mo8上的氧原子也可与pva表面的氧原子形成大量的氢键,降低了(nh4)2{(γ-mo8o26)[vo(cf)2]2}从pva上的释放速率,使得(nh4)2{(γ-mo8o26)[vo(cf)2]2}在水溶液中的溶解度明显降低,因而达到了缓释作用。实验例6样品体外缓释抗菌活性测定本实验例所用样品为实施例2和对比例2制得的样品。复合膜的抑菌活性测试采用纸片法,将复合膜代替纸片来进行实验操作,其中,图5a表示实施例2(s)和对比例2(d)对大肠杆菌的缓释抗菌效果;图5b表示实施例2(s)和对比例2(d)对金黄色葡萄球菌的缓释抗菌效果。由图5a中可知,实施例2和对比例2对于大肠杆菌的抑菌直径分别为27mm和31mm(pva膜无抑菌作用,吸水溶胀后的直径为11mm)。为了进一步对比实施例2和对比例2的抗菌活性,再次进行了三次循环测试。如图5aⅱ所示,在ⅱ中,实施例2和对比例2对于大肠杆菌的抑菌直径分别为31mm和27mm,与ⅰ相比,对比例2的抑菌直径降低了13%,而实施例2的抑菌直径增加了15%;在ⅲ中,对比例2的对于大肠杆菌的抑菌直径与pva薄膜的抑菌直径相同,即为零,而实施例2对大肠杆菌的抑菌直径与ⅱ相比下降19%;在ⅳ中,对实施例2对大肠杆菌的抑菌直径(16nm)与ⅲ相比下降36%,下降趋势相对缓慢。以上结合具体实施方式和范例性实例对本发明进行了详细说明,不过这些说明并不能理解为对本发明的限制。本领域技术人员理解,在不偏离本发明精神和范围的情况下,可以对本发明技术方案及其实施方式进行多种等价替换、修饰或改进,这些均落入本发明的范围内。本发明的保护范围以所附权利要求为准。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1