一种含酚羟基氨基酸-N-硫代羧酸酐单体及其合成和聚合方法与流程
本发明属于高分子合成领域,具体涉及一种含酚羟基氨基酸-n-硫代羧酸酐(nta)单体的合成及聚合方法。
背景技术:
聚氨基酸具有优异的生物相容性和可降解性,被广泛地应用于生物医学领域,如药物载体和组织工程等(j.am.chem.soc.2007,129,5362-5363)。氨基酸-n-羧酸酐(n-carboxyanhydride,简称nca)和氨基酸-n-硫代羧酸酐(n-thiocarboxyanhydride,简称nta)是两类常见的合成聚氨基酸的环状单体。早期的研究发现,羟基具有亲核性,可以引发nca单体聚合(angew.chem.,int.ed.,2006,45,5752-5784)。因此带羟基的nca单体在合成与聚合时需要保护羟基,聚合后需要脱保护。最新研究发现,与nca单体相比,nta单体更为稳定,羟基不能引发其聚合(macromolecules,2017,50,3066-3077)。
氨基酸-n-硫代羧酸酐(包括n-取代氨基酸-n-硫代羧酸酐),是nca单体的硫代类似物。与nca单体相比,其对水和热不敏感,易于储存。同时合成和提纯过程不需要严格的无水无氧操作。有机胺可以引发氨基酸-nta单体的一种或多种进行活性开环聚合,得到均聚、嵌段和多嵌段的聚氨基酸(j.polym.sci.parta:polym.chem.,2017,55,404-410)。氨基酸-nta单体一般可由相应的n-乙氧基硫代羰基氨基酸与三溴化磷反应制备(j.macromol.sci.parta-pureappl.chem.2008,45,425-430)。
目前尚未见侧链带有酚羟基的聚氨基酸的直接合成的报道。
技术实现要素:
本发明的目的是提供一种含酚羟基氨基酸-n-硫代羧酸酐(nta)单体及其合成及聚合方法。该合成方法中,原料廉价易得,合成路线简单,目标产物结构明确。该聚合方法中,聚合反应条件温和,得到的聚氨基酸链长可以通过单体/引发剂的投料比控制,且聚合物结构明确。
所述含酚羟基的氨基酸-n-硫代羧酸酐单体和n-取代甘氨酸-n-硫代羧酸酐单体的结构式分别如下式(1)和(2)所示:
式(1)和式(2)中,n=1~5,r1、r2、r3、r4、r5独立地选自h、羟基、烷基、烷氧基、卤素、三氟甲基、硝基、氰基,可以相同也可以不同。其聚合活性上不存在显著差异。
所述的-ch2-个数(n)优选为1~5,进一步优选为n=1。
式(1)中n=1,当r3为羟基且r1、r2、r4、r5均为氢原子时的氨基酸-n-硫代羧酸酐称为酪氨酸-nta(简称为tyr-nta);当r2、r3为羟基且r1、r4、r5为氢原子时的氨基酸-n-硫代羧酸酐称为多巴-nta(简称为dopa-nta);式(2)中n=1,当r2、r3为羟基且r1、r4、r5为氢原子时的氨基酸-n-硫代羧酸酐称为n-多巴-nta(简称为ndopa-nta)。
作为优选,r1、r2、r3、r4、r5独立地选自h、羟基或c1~c5烷基,且r1~r5中至少一个为羟基。
作为进一步的优选,所述的含酚羟基氨基酸-n-硫代羧酸酐单体为以下化合物中的一种:
本发明还提供了一种含酚羟基氨基酸-n-硫代羧酸酐单体的制备方法,包括以下步骤:
含酚羟基的氨基酸(包括含酚羟基的n-取代甘氨酸)与s-乙氧基硫代羰基巯基乙酸(xaa)在氢氧化钠的水溶液中反应合成对应的n-乙氧基硫代羰基取代氨基酸,后者在有机溶剂中与关环剂反应成环得到含酚羟基的氨基酸-nta单体。
所述关环剂可以是三溴化磷、三氯化磷、五氯化磷、二氯亚砜等。
具体合成路线如下式:
本发明还提供了一种上述单体的聚合方法,包括:在有机胺的引发作用下,含酚羟基的氨基酸-nta单体的一种或者多种在溶剂中发生聚合反应,反应完全之后经过后处理得到侧链带有酚羟基的聚氨基酸。
本发明中聚合反应引发剂为有机胺,其可以为伯胺、仲胺、叔胺、硅基胺中的任意一种,例如正丁胺、苄胺、二乙胺、六甲基二硅氮烷等。同类有机胺的引发效果不存在显著差异。单体与引发剂的摩尔比可在一个较宽的范围内变化,进一步地,本发明所述氨基酸-nta单体与有机胺引发剂的摩尔比为5~100:1,进一步优选在5至50之间。
进一步地,本发明所述聚合反应的温度为0℃~150℃,进一步优选在40℃至80℃之间。
进一步地,本发明所述聚合反应的溶剂为二氧六环、四氢呋喃(thf)、n,n-二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺(dmac)、四甲基脲(tmu)、二甲亚砜(dmso)、环丁砜、硝基苯、乙腈、苯甲腈、n-甲基吡咯烷酮(nmp)、甲苯、二氯甲烷和三氯甲烷中的一种或者多种。
进一步地,本发明所述聚合反应时间为0.5小时~4天,进一步优选为12小时~48小时。
作为更进一步的优选,聚合单体为tyr-nta(即式(1)中的r3为羟基且r1、r2、r4、r5均为氢原子),所述的溶剂为四氢呋喃,所述的引发剂为正丁胺;作为另外的优选,聚合单体为dopa-nta(即式(1)中的r2、r3为羟基且r1、r4、r5为氢原子);作为另外的优选,聚合单体为ndopa-nta(即式(2)中的r2、r3为羟基且r1、r4、r5为氢原子)。得到的聚氨基酸链长可以通过单体与引发剂的投料比控制,且聚合物结构通过核磁氢谱(1hnmr)、基质辅助激光解析/离子化时间飞行质谱(maldi-tofms)等分析方法得以明确表征。
与现有技术相比,本发明的有益效果是:(1)首次合成含有酚羟基的氨基酸(包括含酚羟基的n-取代甘氨酸)-n-硫代羧酸酐(nta)单体,在此之前此类单体从未经报道。(2)对于所述氨基酸-nta单体的酚羟基不需要保护,可直接进行开环聚合,得到了侧链带有酚羟基的聚氨基酸产物;而现有技术中,带酚羟基的氨基酸-n-羧酸酐(nca)单体在合成与聚合时需要保护羟基,聚合后需要脱保护。其单体合成难度、聚合难度与本发明的氨基酸-nta单体合成及聚合相距甚远。本发明提供的含酚羟基氨基酸-n-硫代羧酸酐(nta)单体的聚合方法具有如下特性:(i)引发剂的原料廉价易得,具有很高的引发活性;(ii)聚合条件温和,可制备得到较高分子量的聚氨基酸,聚合度可达100及以上;(iii)制备所得聚氨基酸的分子量可以通过单体与引发剂的摩尔比进行调节,投料比在5~100之间;(vi)可以与多种不同类型的氨基酸-nta单体共聚合,得到结构、功能不同的聚氨基酸。(3)本发明所用的氨基酸-nta单体具有如下特性:性质稳定,合成过程简单易行,能够长时间保存,可以实现大批量的工业化生产。(4)本发明所得的产物聚氨基酸具有良好的生物相容性和生物降解性,无毒,是一种优良的生物材料。
附图说明
图1为单体tyr-nta、单体dopa-nta的核磁氢谱图。
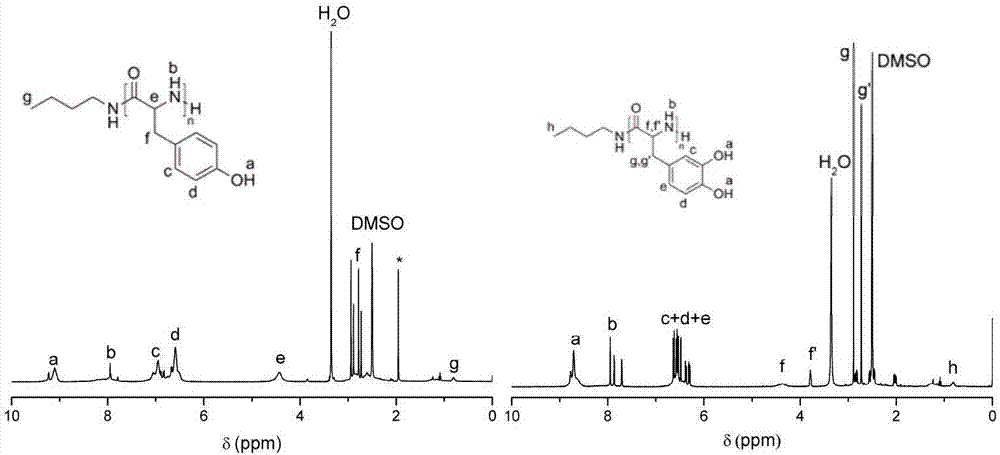
图2为实施例1、实施例4正丁胺分别引发tyr-nta、dopa-nta均聚合的聚氨基酸产物的核磁氢谱图。
图3为实施例1、实施例4正丁胺分别引发tyr-nta、dopa-nta均聚合的聚氨基酸产物的maldi-tof质谱图。
具体实施方式
以下结合具体实施将对本发明进行进一步的说明。
所得聚氨基酸的分子量和结构采用1hnmr和maldi-tofms测定。聚合物的绝对分子量用1hnmr端基法标定,核磁在brukeravancedmx400(1h:400mhz)仪器上测定,用氘代二甲亚砜(dmso-d6)作为溶剂,四甲基硅烷(tms)作为内标。聚合物的结构用maldi-tofms分析,在brukerultraflexmaldi-tof质谱仪上测定。氮激光波长设定为337nm,脉冲间隔为3ns。在反射模式下测定,加速电压为20kv。以2,5-二羟基苯甲酸(dhb)为基质。
实施例1
在反应瓶中分别加入36.2g(0.20mol)酪氨酸、32.0g(0.80mol)氢氧化钠、36.0g(0.20mol)s-乙氧基硫代羰基巯基乙酸(xaa),用150ml去离子水溶解,室温下搅拌反应72h合成得到31.3gn-乙氧基硫代羰基取代酪氨酸(0.12mol,收率为58.2%)。后者在乙酸乙酯中与pbr3反应成环得到9.63gtyr-nta单体(0.043mol,收率为35.7%)。
在反应瓶中加入0.223g(1.00mmol)的tyr-nta,用2.0ml四氢呋喃(thf)溶解,再加入0.12ml正丁胺的thf溶液(0.170mol/l,0.020mmol),tyr-nta与正丁胺的摩尔比为50:1,密封后置于40℃油浴中反应4天,反应后将混合物倒入乙醚中沉淀、过滤,将所得的聚合物真空干燥1天,得到聚酪氨酸,收率为94.2%。所得聚合物数均绝对分子量为5.3kda,分子量分布为1.15。得到的聚合物的核磁氢谱图如图2左侧所示,maldi-tof质谱图如图3d,图3e和图3f所示。
实施例2
其他聚合条件与实施例1相同,所不同的是用苄胺作为引发剂引发tyr-nta开环聚合,溶剂为n-甲基吡咯烷酮(nmp),tyr-nta与苄胺的摩尔比为20:1,80℃恒温槽中反应12h,所得聚酪氨酸,收率为85.8%。所得聚合物数均绝对分子量为3.0kda。
实施例3
其他聚合条件与实施例1相同,所不同的是用分子量2000的端氨基聚乙二醇(peg-nh2)引发tyr-nta开环聚合,溶剂为n,n-二甲基乙酰胺(dmac),tyr-nta与peg-nh2的摩尔比为10:1,0℃恒温槽中反应2天,所得聚氨基酸收率为99.0%。所得共聚物数均绝对分子量为3.7kda。
实施例4
在反应瓶中分别加入19.7g(0.10mol)多巴、12.0g(0.30mol)氢氧化钠、18.0g(0.20mol)s-乙氧基硫代羰基巯基乙酸(xaa),用150ml去离子水溶解,室温下搅拌反应72h合成得到25.8gn-乙氧基硫代羰基取代多巴(0.09mol,收率为89.9%)。后者在乙酸乙酯中与pbr3反应成环得到6.52gdopa-nta单体(0.027mol,收率为30.3%)。
在反应瓶中加入0.387g(1.62mmol)的dopa-nta,用2.9ml四氢呋喃(thf)溶解,再加入0.38ml正丁胺的thf溶液(0.206mol/l,0.078mmol),dopa-nta与正丁胺的摩尔比为20:1,密封后置于100℃油浴中反应4h,反应后将混合物倒入乙醚中沉淀、过滤,将所得的聚合物真空干燥1天,得到聚多巴,收率为82.8%。所得聚合物数均绝对分子量为3.1kda,分子量分布为1.24。得到的聚合物的核磁氢谱图如图2右侧所示,maldi-tof质谱图如图3a,图3b和图3c所示。
实施例5
其他聚合条件与实施例4相同,所不同的是用苄胺作为引发剂引发dopa-nta开环聚合,溶剂为n-甲基吡咯烷酮(nmp),dopa-nta与苄胺的摩尔比为10:1,80℃恒温槽中反应12h,所得聚多巴,收率为94.4%。所得聚合物数均绝对分子量为1.7kda。
实施例6
其他聚合条件与实施例4相同,所不同的是用分子量2000的端氨基聚乙二醇(peg-nh2)引发dopa-nta开环聚合,溶剂为n,n-二甲基乙酰胺(dmac),dopa-nta与peg-nh2的摩尔比为5:1,0℃恒温槽中反应2天,所得聚氨基酸收率为98.4%。所得共聚物数均绝对分子量为2.9kda。
实施例7
在反应瓶中加入0.452g(1.89mmol)的ndopa-nta,用3.6ml四氢呋喃(thf)溶解,再加入0.18ml正丁胺的thf溶液(0.106mol/l,0.019mmol),ndopa-nta与正丁胺的摩尔比为100:1,密封后置于60℃油浴中反应1天,反应后将混合物倒入乙醚中沉淀、过滤,将所得的聚合物真空干燥1天,得到聚-n-多巴,收率为70.2%。所得聚合物数均绝对分子量为10.8kda。
- 还没有人留言评论。精彩留言会获得点赞!
- 苯甲酰胺类,含有苯甲酰胺类的组合物以及苯甲酰胺类的制备方法与流程
- 一种硫酰胺的加工工艺的制作方法与工艺
- 一种硫酰胺的制备方法与流程
- 含苯甲酰胺类、二硝基苯胺类、二硫代氨基甲酸酯类杀菌剂和生防菌的组合物、制剂及应用的制作方法与工艺
- 含羧酸酰胺类、苯甲酰胺类杀菌剂和生防菌的菌药组合物、制剂及应用的制作方法与工艺
- 一种N‑烷基羟肟酸‑二硫代氨基甲酸酯捕收剂、制备及其应用的制作方法与工艺
- 含酰胺键的1,3,4‑噁二唑硫/氧醚类化合物及其制备方法及应用与流程
- 来自羟基官能的缩酮酸、酯或酰胺的高再循环含量的聚酯型多元醇的制造方法与工艺
- N‑羟基硫代琥珀酰亚胺生物素酯及其衍生物的制备方法与流程
- 包含(硫代)磷酸三酰胺和其它化合物如胺和着色剂的具有改进脲酶抑制效果的组合物的制造方法与工艺