苯乙醇胺类β受体激动剂的合成方法与流程
本发明涉及化工技术领域,特别是涉及一种苯乙醇胺类β受体激动剂的合成方法。
背景技术:
随着人类生活水平的不断提高,人们对食品安全的关注度越来越高,然而,食品安全问题不容乐观,特别是动物源性食品安全问题,已成为全球的议题,而兽药残留是导致动物源性食品安全问题的主要原因。兽药残留是化学性风险的一种,这种化学性风险不是消费者能够用感官来识别的,因此加强对兽药残留的检测,对保护人类身体健康有着重要的意义。
β受体激动剂临床上可用于治疗人和动物的哮喘类似病症,在化学结构上类似肾上腺素的苯乙醇胺类药物。另外,将其添加于饲料中,可以增加动物瘦肉量,减少饲料使用,使肉品早上市,因此这类化合物俗称“瘦肉精”。但其易在动物体内残留。人食用含β受体激动剂较高的食物后,会出现急性中毒的症状如:恶心,呕吐,神经过敏,肌肉振颤,心率加快等,甚至造成生命危险。近年来被检测到的瘦肉精类β受体激动剂有溴布特罗,克伦特罗,西马特罗及其盐等,严重危害着畜产品的安全。我国农业部1997年发布公告所有可食用动物组织中不得检测出此类药物,禁止瘦肉精应用于畜牧业。
目前关于β受体激动剂合成方法公开文献较少,而且操作复杂,成本较高,且原子利用率较低,而且难以使用通用路线得到多种苯乙醇胺类化合物,针对现有合成路线的不足,有必要提供一种路线简单、原料廉价,且原子利用率较高的通用的合成多种苯乙醇胺类化合物的方法,合成出的产物可以作为测定β受体激动剂的标准试剂,满足我国食品安全的检测需求。
技术实现要素:
本发明所要解决的技术问题是提供一种苯乙醇胺类β受体激动剂的合成方法,路线简单、原料廉价,且原子利用率较高。
本发明为解决上述技术问题而采用的技术方案是提供一种苯乙醇胺类β受体激动剂的合成方法,所述苯乙醇胺类β受体激动剂如通式(ⅳ)所示:
所述β受体激动剂类化合物为:
克伦特罗:r1=cl,r2=cl,r=叔丁胺
溴布特罗:r1=br,r2=br,r=叔丁胺
西布特罗:r1=h,r2=cn,r=叔丁胺
西马特洛:r1=h,r2=cn,r=异丙胺;或
溴氯布特罗:r1=br,r2=cl,r=叔丁胺
包括以下步骤:s1:将4-氨基苯乙酮溶解在有机溶剂中,与亲电取代试剂发生苯环上的卤代反应,生成卤苯类中间体;所述卤苯类中间体在有机溶剂或水中,在金属催化剂的催化下与氰化试剂发生亲核取代反应,生成如通式(ⅰ)所示苯乙酮类中间体;s2:通式(ⅰ)所示苯乙酮类中间体在有机溶剂中,与溴化铜发生羰基α溴代反应生成通式(ⅱ)所示α-溴代苯乙酮类中间体;s3:通式(ⅱ)所示α-溴代苯乙酮类中间体在有机溶剂中与叔丁胺或异丙胺反应生成如通式(ⅲ)所示苯乙酮胺类中间体;s4:通式(ⅲ)所示苯乙酮胺类中间体在有机溶剂中,与还原氢化试剂反应生成苯乙醇胺类β受体激动剂;
通式(ⅰ)(ⅱ)(ⅲ)中r1、r2分别独立选自氢、氯、溴和腈基中的一种,r为叔丁胺或异丙胺。
进一步地,所述步骤s1中4-氨基苯乙酮与亲电取代试剂的摩尔比为1:(1~2.5),反应温度为0℃~40℃,反应时间为0.5~5小时;卤苯类中间体与金属催化剂和氰化试剂的摩尔比为1:(0.01~0.05):(0.3~0.8),反应温度为80~120℃,反应时间为4~24小时。
进一步地,所述步骤s1中所用的亲电取代试剂选自:nbs、ncs、br2和cl2中的至少一种;氰化试剂选自:亚铁氰化钠盐、亚铁氰化钾盐或其水合物中的一种;金属催化剂为四三苯基膦钯。
进一步地,所述步骤s1中4-氨基苯乙酮与亲电取代试剂的投料比不同所得的主产物和副产物都可以作为合成不同苯乙醇胺类β受体激动剂的原料。
进一步地,所述步骤s1中,还加入dbu,卤苯类中间体与dbu的摩尔比为1:(0.25-0.5)。
进一步地,所述步骤s2中,苯乙酮类中间体与溴化铜的摩尔比为1:1.5~2.5;反应温度为55℃~75℃,反应时间为2~3小时。
进一步地,所述步骤s3中,α-溴代苯乙酮类中间体与叔丁胺或异丙胺的摩尔比为1:1.5~2.5,反应温度为0℃~60℃,反应时间为2~5小时。
进一步地,所述步骤s4中,苯乙酮胺类中间体与还原氢化试剂的摩尔比为1:1~2.5,反应温度为0℃~40℃,反应时间为6~16小时。
进一步地,所述步骤s4所用的还原氢化试剂选自:硼氢化钠,硼氢化钾和氢化铝锂中的至少一种。
进一步地,所述步骤s1-s4所用的有机溶剂选自:乙腈,乙酸乙酯,二氯甲烷,乙醇,四氢呋喃,甲醇,氯仿和叔丁醇中的至少一种。
本发明对比现有技术有如下的有益效果:本发明提供的苯乙醇胺类β受体激动剂的合成方法简单高效且原料廉价易得原子利用率较高,通过亲电取代、亲核取代、还原反应即可制备得到苯乙醇胺类β受体激动剂。本发明可用于合成多种苯乙醇胺类β受体激动剂,合成产物化学纯度大于99%,可充分满足食品安全领域“瘦肉精”等禁用兽药残留检测及其代谢机理研究。
附图说明
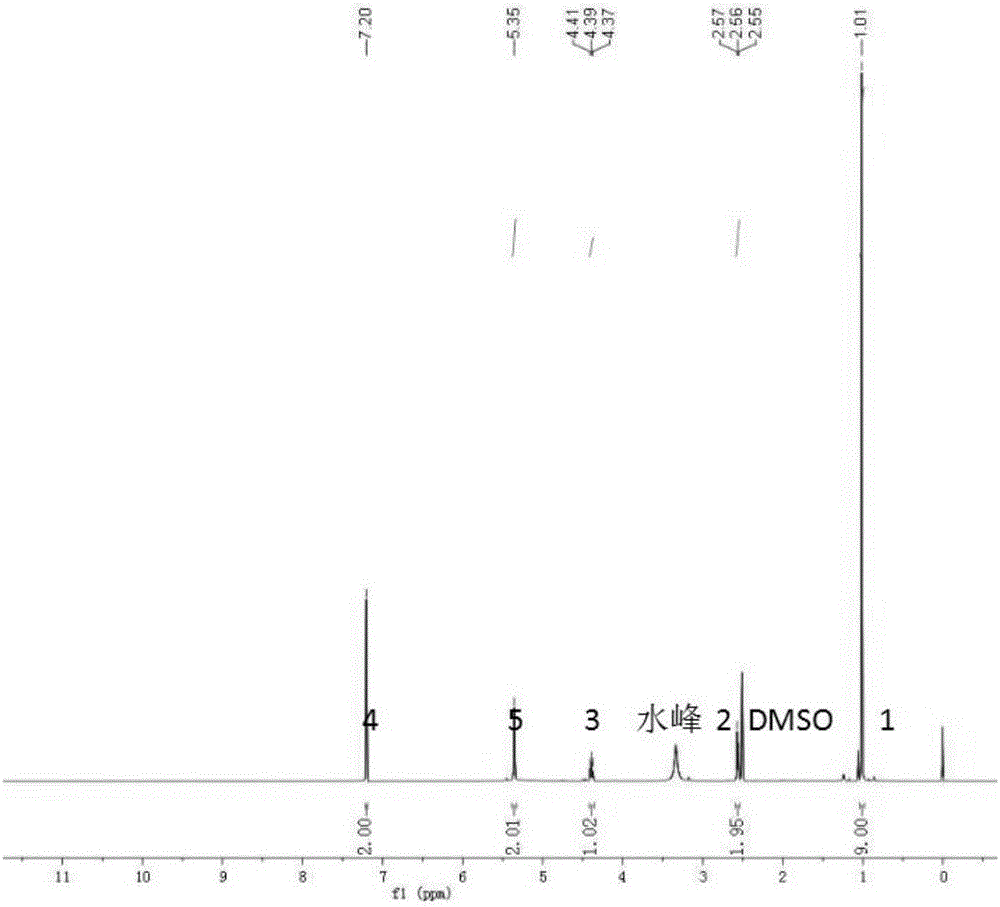
图1为克伦特罗的核磁共振氢谱图;以dmso为溶剂,通过bruke-400m核磁共振仪器获得。
图2为溴布特罗的核磁共振氢谱图;以cd3od为溶剂,通过bruke-400m核磁共振仪器获得。
图3为西布特罗的核磁共振氢谱图;以cd3od为溶剂,通过bruke-400m核磁共振仪器获得。
图4为西马特罗的核磁共振氢谱图;以cd3od为溶剂,通过bruke-400m核磁共振仪器获得。
图5为溴氯布特罗的核磁共振氢谱图;以cd3od为溶剂,通过bruke-400m核磁共振仪器获得。
图6为克伦特罗的高效液相色谱图。
图7为溴布特罗的高效液相色谱图。
图8为西布特罗的高效液相色谱图。
图9为西马特罗的高效液相色谱图。
图10为溴氯布特罗的高效液相色谱图。
具体实施方式
下面结合附图和实施例对本发明作进一步的描述,但不应理解为是对本发明的限制。
实施例1
4-氨基-3,5-二氯苯乙酮(克伦特罗)的合成方法
称取7.9g(59.2mmol)ncs于250ml三口烧瓶,加入60mlmecn,搅拌溶解,用恒压滴液漏斗将溶解在50ml乙腈中的4-氨基苯乙酮(4g,29.6mmol)缓慢滴入反应装置,半小时滴加完毕,室温下搅拌3小时。旋蒸出大部分溶剂,加入乙酸乙酯搅拌溶解,每次用50ml纯水洗3次,收集有机相,无水硫酸钠干燥。过滤后旋干溶剂得到主产物苯乙酮中间体4-氨基-3,5-二氯苯乙酮4.8g,产率为80%;同时得到副产物4-氨基-3-氯苯乙酮0.9g,产率为18%,可用于实施例5作为制备溴氯布特罗的原料。
从4-氨基-3,5-二氯苯乙酮合成4-氨基-3,5-二氯溴代苯乙酮
称取4.8g(23.7mmol)的4-氨基-3,5-二氯苯乙酮于250ml圆底烧瓶,加入60mlea(乙酸乙酯),60mlchcl3和20mlch3ch2oh溶剂,回流温度下,搅拌溶解。称取7.8g(35mmol)溴化铜,加入反应装置中,共反应2h。趁热抽滤,每次用20ml的ch2cl2洗涤滤饼3次,将滤液转入分液漏斗中,用3次50ml纯水洗涤滤液,收集有机相,加入无水硫酸钠干燥。过滤后旋干溶剂,用5.0g质量百分比浓度75%的乙醇重结晶得到4-氨基-3,5-二氯溴代苯乙酮。
从4-氨基-3,5-二氯溴代苯乙酮合成克伦特罗
称取5.0g(17.8mmol)的4-氨基-3,5-二氯溴代苯乙酮于250ml圆底烧瓶,加入50ml四氢呋喃(thf)和50ml乙醇(ch3ch2oh),置于冰水浴中,搅拌溶解,装置除氧,n2气保护,20min后用注射器吸取3.8ml(35.6mmol)的叔丁胺,缓慢加入反应装置,在0℃下继续反应3h,常温反应1h。
然后将反应装置置于冰水浴,称取1.5g(28mmol)的kbh4缓慢加入反应装置,2h后,撤去冰浴,量取50ml甲醇(ch3oh)加入反应装置,常温下搅拌16h,旋蒸除去大部分溶剂,加入30ml水淬灭反应,二氯甲烷萃取三次。合并有机相后用无水硫酸钠干燥。过滤后除去溶剂,通过柱层析得到1.7g克伦特罗,产率为35%。
图1为克伦特罗的核磁共振氢谱图;以dmso为溶剂,通过bruke-400m核磁共振仪器获得。化学位移δ7.20(2h,s),5.35(2h,s),4.41-4.37(1h,m),2.57-2.55(2h,m),1.01(9h,s)。
图6为克伦特罗的高效液相色谱图,样品溶于流动相,以乙腈:0.01mkh2po4(ph=3)=20:80为流动相,1.0ml/min的流速通过柱温为30℃的液相柱(cnwathenac18-wp4.6*250mm,5um(laeq-462572)),通过dad(213nm)检测器获得克伦特罗的高效液相色谱图,由图6可知,样品纯度达到99%以上。
实施例2
4-氨基-3,5-二溴苯乙酮(溴布特罗)的合成方法
称取11.6g,65.1mmoln-溴代丁二酰亚胺(nbs)于250ml三口烧瓶,加入60mlmecn,搅拌溶解,用恒压滴液漏斗将溶解在50ml乙腈中的4g,29.6mmol4-氨基苯乙酮缓慢滴入反应装置,半小时滴加完毕,室温下搅拌3小时。旋蒸出大部分溶剂,加入乙酸乙酯搅拌溶解,用50ml纯水洗3次,收集有机相,无水硫酸钠干燥。过滤后旋干溶剂得到6.9g苯乙酮中间体主产物4-氨基-3,5-二溴苯乙酮,产率为80%;同时得到1.2g副产物4-氨基-3-溴苯乙酮,产率为20%,可用于实施例3和4作为制备西布特罗和西马特洛的原料。
从4-氨基-3,5-二溴苯乙酮合成4-氨基-3,5-二溴溴代苯乙酮
称取6.9g(23.7mmol)4-氨基-3,5-二溴苯乙酮于250ml圆底烧瓶,加入60mlea,60mlchcl3和20mlch3ch2oh溶剂,回流温度下,搅拌溶解。称取10.5g(47.4mmol)溴化铜,加入反应装置中,共反应2h。趁热抽滤,每次用20mlch2cl2洗涤滤饼3次,将滤液转入分液漏斗中,每次用50ml纯水洗涤滤液3次,收集有机相,加入无水硫酸钠干燥。过滤后旋干溶剂,乙醇重结晶得到6.3g4-氨基-3,5-二溴溴代苯乙酮,产率为71%。
从4-氨基-3,5-二溴溴代苯乙酮合成溴布特罗
称取6.3g(16.8mmol)4-氨基-3,5-二溴溴代苯乙酮于250ml圆底烧瓶,加入50mlthf和50mlch3ch2oh溶剂中,置于冰水浴中,搅拌溶解,装置除氧,n2气保护,20min后用注射器吸取2.7ml(25.2mmol)叔丁胺,缓慢加入反应装置,在0℃下继续反应3h,常温反应1h。
然后将反应装置置于冰水浴,称取1.5g(28mmol)kbh4缓慢加入反应装置,2h后,撤去冰浴,量取50mlch3oh加入反应装置,常温下搅拌16h,旋蒸除去大部分溶剂,加入30ml水淬灭反应,二氯甲烷萃取三次。合并有机相后用无水硫酸钠干燥。过滤后除去溶剂,通过柱层析得到2.0g溴布特罗,产率为33%。
图2为溴布特罗的核磁共振氢谱图;以cd3od为溶剂,通过bruke-400m核磁共振仪器获得。化学位移δ7.43(2h,s),4.57-4.54(1h,m),2.74-2.64(2h,m),
1.15(9h,s)。
图7为溴布特罗的高效液相色谱图,检测方法和克伦特罗相似,由图7可知,样品纯度达到99%以上。
实施例3
4-氨基-3-溴苯乙酮(西布特罗)的合成方法
称取4g(29.6mmol)4-氨基苯乙酮于250ml三口烧瓶,加入40mlmecn,搅拌溶解,用恒压滴液漏斗将溶解在50ml乙腈中的5.8g(32.6mmol)nbs缓慢滴入反应装置,半小时滴加完毕,室温下搅拌3小时。旋蒸出大部分溶剂,加入乙酸乙酯搅拌溶解,每次用50ml纯水洗3次,收集有机相,无水硫酸钠干燥。过滤后旋干溶剂得到苯乙酮中间体主产物4-氨基-3溴苯乙酮4.5g,产率为71%;同时得到1.2g副产物4-氨基-3,5-二溴苯乙酮,产率为14%,可用于实施例2作为合成溴布特罗的原料。
从4-氨基-3-溴苯乙酮合成4-氨基-3-氰基苯乙酮
称取4.5g(21mmol)4-氨基-3-溴苯乙酮,3.6g(8.5mmol)亚铁氰化钾三水合物,0.45g(0.42mmol)四三苯基膦钯于三口瓶中,量取35ml叔丁醇,35ml纯水加入反应装置,用注射器加入0.8ml1,8-二氮杂双环[5.4.0]十一碳-7-烯(dbu),反应装置除氧后n2保护下,回流温度下,反应16h。反应结束后抽滤,收集滤液,每次用20ml二氯甲烷萃取3次,收集有机相,加入无水硫酸钠干燥,抽滤旋干得到4-氨基-3-氰基苯乙酮2.8g,产率为85%。
从4-氨基-3-氰基苯乙酮合成4-氨基-3-氰基溴代苯乙酮
称取2.8g(17.5mmol)4-氨基-3-氰基苯乙酮于250ml圆底烧瓶,加入60mlea、60mlchcl3和20mlch3ch2oh溶剂,回流温度下,搅拌溶解。称取7.8g(35mmol)溴化铜,加入反应装置,回流反应2h后,趁热抽滤,收集滤液,每次20ml水洗3次,收集有机相,加入无水硫酸钠干燥,抽滤旋干溶剂得到4-氨基-3-氰基溴代苯乙酮3.4g,产率为80%。
从4-氨基-3-氰基溴代苯乙酮合成西布特罗
称取3.4g(14mmol)4-氨基-3-氰基溴代苯乙酮于250ml圆底烧瓶,加入50mlthf和50mlch3ch2oh溶剂,置于冰水浴中,搅拌溶解,20min后用注射器吸取3ml(28mmol)叔丁胺,缓慢加入反应装置,在0℃下继续反应3h,常温反应1h。
然后将反应装置置于冰水浴,称取1.5g(28mmol)kbh4缓慢加入反应装置,2h后,撤去冰浴,量取50mlch3oh加入反应装置,常温下搅拌16h(过夜反应即可),旋蒸除去大部分溶剂,加入30ml水淬灭反应,二氯甲烷萃取三次。合并有机相后用无水硫酸钠干燥。过滤后除去溶剂,通过柱层析得到1.0g西布特罗,产率为30%。
图3为西布特罗的核磁共振氢谱图;以cd3od为溶剂,通过bruke-400m核磁共振仪器获得。化学位移δ7.38-7.34(2h,m),6.83(1h,d),4.58(1h,dd),2.72(2h,ddd),1.16(9h,s)。
图8为西布特罗的高效液相色谱图,检测方法和克伦特罗相似,由图8可知,样品纯度达到99%以上。
实施例4
合成4-氨基-3-氰基溴代苯乙酮(西马特洛)的路线与实施例3合成方法相同。
从4-氨基-3-氰基溴代苯乙酮合成西马特罗
称取3.4g(14mmol)4-氨基-3-氰基溴代苯乙酮于250ml圆底烧瓶,加入50mlthf和50mlch3ch2oh的溶剂,置于冰水浴中,搅拌溶解,20min后用注射器吸取3ml(28mmol)异丙胺,缓慢加入反应装置,在0℃下继续反应3h,常温反应1h。
然后将反应装置置于冰水浴,称取1.5g(28mmol)kbh4缓慢加入反应装置,2h后,撤去冰浴,量取50mlch3oh加入反应装置,常温下搅拌16h(过夜反应即可),旋蒸除去大部分溶剂,加入30ml水淬灭反应,二氯甲烷萃取三次。合并有机相后用无水硫酸钠干燥。过滤后除去溶剂,通过柱层析得到1.0g西马特罗,产率为30%。
图4为西马特罗的核磁共振氢谱图;以cd3od为溶剂,通过bruke-400m核磁共振仪器获得。化学位移δ7.37-7.33(2h,m),6.83(1h,d),4.63(1h,dd),2.89(1h,dd),2.78-2.68(2h,m),1.11(6h,dd)。
图9为西马特罗的高效液相色谱图,检测方法和克伦特罗相似,从图9可知,样品纯度达到99%以上。
实施例5
4-氨基-3-氯苯乙酮(溴氯布特罗)的合成方法
称取5g(37mmol)4-氨基苯乙酮于250ml圆底烧瓶,加入60mlmecn,搅拌溶解,然后称取5.4g(40.4mmol)ncs溶解在50mlmecn中,用恒压滴液漏斗将其缓慢滴入反应装置,半小时滴加完毕,室温下搅拌4h。旋蒸出大部分溶剂,加入乙酸乙酯搅拌溶解,每次用50ml纯水洗3次,收集有机相,无水硫酸钠干燥。过滤后旋干溶剂得到3g苯乙酮中间体主产物4-氨基-3-氯苯乙酮,产率为45%;同时得到1.5g,副产物4-氨基-3,5-二氯苯乙酮,产率为20%,可用于实施例1作为合成克伦特罗的原料。
从4-氨基-3-氯苯乙酮合成4-氨基-3-溴-5-氯苯乙酮
称取3g(17.6mmol)4-氨基-3-氯苯乙酮于250ml圆底烧瓶,加入50mlmecn,搅拌溶解,然后称取3.5g(40.4mmol)nbs溶解在50mlmecn中,用恒压滴液漏斗将其缓慢滴入反应装置,半小时滴加完毕,室温下搅拌1.5h,旋蒸出大部分溶剂,加入乙酸乙酯搅拌溶解,每次用50ml纯水洗3次,收集有机相,无水硫酸钠干燥。过滤后旋干溶剂得到3.5g苯乙酮中间体4-氨基-3-溴-5-氯苯乙酮,产率为80%。
从4-氨基-3-溴-5-氯苯乙酮合成4-氨基-3-溴-5-氯溴代苯乙酮
称取3.5g(17.6mmol)4-氨基-3-溴-5-氯苯乙酮于250ml圆底烧瓶,加入60mlea、60mlchcl3和20mlch3ch2oh溶剂,回流温度,搅拌溶解。称取6.5g(29mmol)溴化铜,回流反应2h。趁热抽滤,每次用20mlch2cl2洗涤滤饼3次,将滤液转入分液漏斗中,每次用50ml纯水洗涤滤液3次,收集有机相,加入无水硫酸钠干燥。过滤后旋干溶剂,乙醇重结晶得到4-氨基-3-溴-5-氯溴代苯乙酮3.5g,产率为75%。
从4-氨基-3-溴-5-氯溴代苯乙酮合成溴氯布特罗
称取3.5g(10.7mmol)4-氨基-3-溴-5-氯溴代苯乙酮于250ml圆底烧瓶,加入30mlthf和30mlch3ch2oh的溶剂,置于冰水浴中,搅拌溶解,20min后用注射器吸取2.3ml(22mmol)叔丁胺,缓慢加入反应装置,反应装置除氧n2保护,在0℃下继续反应3h,常温反应1h。
然后将反应装置置于冰水浴,称取1.19g(22mmol)kbh4缓慢加入反应装置,2h后,撤去冰浴,量取30mlch3oh加入反应装置。加入20ml纯水淬灭反应,旋蒸出大部分溶剂,剩余部分转入分液漏斗中,每次用50mlch2cl2:h2o(体积比2:1)洗3次,收集有机相,加入无水硫酸钠干燥。通过柱层析得到1.3g溴氯布特罗,产率为37%。
图5为溴氯特罗的核磁共振氢谱图;以cd3od为溶剂,通过bruke-400m核磁共振仪器获得。化学位移δ7.36-7.35(1h,m),7.24-7.23(1h,m),4.53-4.51(1h,m),2.71-2.59(2h,m),1.12-1.11(9h,m)。
图10为溴氯布特罗的高效液相色谱图,检测方法和克伦特罗相似,从图10可知,样品纯度达到99%以上。
虽然本发明已以较佳实施例揭示如上,然其并非用以限定本发明,任何本领域技术人员,在不脱离本发明的精神和范围内,当可作些许的修改和完善,因此本发明的保护范围当以权利要求书所界定的为准。
- 还没有人留言评论。精彩留言会获得点赞!