本发明属于医药技术领域,具体涉及一种雷西纳德的精制方法。
背景技术:
雷西纳德,分子式:c17h14brn3o2s,其中文命名:2-((5-溴-4-(4-环丙基萘-1-基)-4h-1,2,4-三氮唑-3-基)硫代)乙酸,商品名:zurampic,其结构式为:
雷西纳德是阿斯利康开发,全球首个上市的尿酸盐重吸收转运子(urati)抑制剂。分别于2015年底、2016年初获得美国fda和欧盟ema的上市批准,联合黄嘌呤氧化酶抑制剂(xoi)用于治疗高尿酸血症相关的痛风;在中国,雷西纳德至今只有1家进口,申报临床,至今没有国内厂家申报,属于3.1类仿制药。
该药物申请专利涉及到合成工艺、晶型、晶型制备方法等诸多方面,目前已知晶型为ⅰ晶型、ⅱ晶型,相对于ⅰ晶型,ⅱ晶型为临床使用晶型。在已见于专利文献报道的合成路线中,对每步中间体及成品的精制步骤,一些环节工序操作起来繁琐,存在不适应工业化大生产要求等缺陷。
技术实现要素:
针对上述现有技术存在的问题,本发明以4-(4-环丙基萘-1-基)-1h-1,2,4-三唑-5(4h)-硫醇为起始原料,经与溴乙酸乙酯烷基化反应、与n-溴代丁二酰亚胺溴代反应、经氢氧化钠水解、氢溴酸中和、精制、干燥,成功的合成出高纯度lesinurad原料药成品,在已存在专利报告其合成路线的基础上,本专利通过对产品和中间体的制备进行进一步完善,在提高收率的基础上,寻找出了在符合工业经济效益基础上得到高纯度的原料药的方法,该过程使用到的溶剂廉价,方法简单,易于进行产品收集,适合工业化扩大生产。
本发明雷西纳德的精制方法涉及的合成路线如下:
本发明所述的一种雷西纳德的精制方法,将中间体c加入纯化水中,然后加入氢溴酸调节ph,具体调节方式重复步骤a)~c),直至检测水相ph在5.0~5.3之间:
a)调节ph值为13~6、加入乙酸乙酯萃取、静止分液、检测水相ph值;
b)调节ph值为6~5、加入乙酸乙酯萃取、静止分液、检测水相ph值;
c)调节ph值为5~3、加入乙酸乙酯萃取、静止分液、检测水相ph值;
收集水相ph在6~5之间的乙酸乙酯相;减压蒸馏出部分溶剂至大量固体析出,沉淀,过滤得到雷西纳德成品;其中所述中间体c的结构如下:
发明人发现,水相ph在6~5之间的乙酸乙酯相含雷西纳德的纯度更高,可达99%以上。而ph值13~6和5~3阶段下的有机相不仅含雷西纳德量很少,并且杂质占比更多。
为了进一步提高收率和纯度,本发明提供一种更为具体的雷西纳德的精制方法,以4-(4-环丙基萘-1-基)-1h-1,2,4-三唑-5(4h)-硫醇为原料,采用烷基化反应、溴代反应、水解、中和、精制合成雷西纳德成品;其中:
1)原料烷基化得中间体a粗品,于乙酸乙酯中,升温至70~80℃,维持该温度使其完全溶解,缓慢降至0~20℃,保温搅拌,抽滤,得中间体a精品;通过本发明所述方法一方面可以实现中间体a粗品的纯化,另一方面,相对于现有的直接从水中析出干燥的方法,使产品更容易干燥,大大缩短了干燥时间,便于第二步要求严格的无水环境的反应;
2)中间体a精品经溴代反应得到中间体b的反应液,于0~20℃通过向反应液中加入反应液体积量1~4倍的纯化水,使中间体b从体系中析出,抽滤得到中间体b粗品;将中间体b粗品加入到纯化水、乙酸乙酯与10%nabr的混合溶液中,20℃~30℃下,搅拌溶解,静置,分液,收集有机相,经减压蒸馏,重结晶,沉淀,过滤得第二步中间体b精品;
3)中间体b精品水解得中间体c,将中间体c加入纯化水中,然后加入氢溴酸调节ph,具体调节方式重复步骤a)~c),直至检测水相ph在5.0~5.3之间:a)调节ph值为13~6、加入乙酸乙酯萃取、静止分液、检测水相ph值;b)调节ph值为6~5、加入乙酸乙酯萃取、静止分液、检测水相ph值;c)调节ph值为5~3、加入乙酸乙酯萃取、静止分液、检测水相ph值;收集水相ph在6~5之间的乙酸乙酯相,收集的乙酸乙酯相,减压蒸馏出部分溶剂至大量固体析出,沉淀,过滤得到雷西纳德成品。
进一步的,步骤1)中保温搅拌1.5-2.5h;抽滤后优选用正庚烷和乙酸乙酯的混合溶液洗涤沉淀,再抽滤;所述混合溶液中正庚烷和乙酸乙酯的体积比优选为1:0.5~3。
进一步的,步骤2)中混合溶液中纯化水、乙酸乙酯与10%nabr的体积比为:15~2:15~2:1,若10%nabr超过此比例,则过多的nabr会使最终产品里nabr残留而造成炽灼残渣不合格。
进一步的,步骤2)中重结晶所用的溶剂优选为四氢呋喃与正庚烷的混合溶液,混合溶液中四氢呋喃与正庚烷的体积比为(1~3):1。
进一步的,步骤2)中重结晶用溶剂溶解沉淀时的溶液温度为40℃~70℃,优选50℃~70℃。
进一步的,步骤2)中向反应液中加入纯化水的体积优选为反应液体积量2~4倍。
进一步的,步骤2)中得到中间体b的具体方法为:将中间体a精品加入到四氢呋喃中,10℃~30℃下,搅拌溶解形成溶液,向溶液中分批次加入粉末状n-溴代丁二酰亚胺,10℃~30℃下,持续搅拌7~12小时,向药液中滴加入3%的焦亚硫酸钠水溶液及纯化水至总加入水体积为反应溶液体积的1~4倍,搅拌至大量固体析出,沉淀,过滤得第二步中间体b粗品。通过加入3%的焦亚硫酸钠,可除去未反应完全的nbs及nbs生产的溴,不使用会使终产品中带有粉红色。优选的,3%的焦亚硫酸钠的加入量为中间体a精品质量的1~4倍。
进一步的,步骤3)中收集的乙酸乙酯相得到雷西纳德成品的具体方法为:乙酸乙酯相20~25℃下,减压蒸馏至体系由澄清变浑浊,关闭减压,升温至40±2℃,保温搅拌1.5~2.5小时,降温至20~25℃,继续减压蒸馏至固体与乙酸乙酯的质量体积比为3.5~4,关闭减压,升温至40±2℃,滴加正庚烷,降温至0~5℃,搅拌1.5~2.5小时,抽滤,用正庚烷和乙酸乙酯的混合溶液洗涤沉淀,抽滤干燥得雷西纳德。
进一步地,正庚烷加入的体积量为步骤3)雷西纳德理论质量的0.5~1.5倍。
进一步的,所用的正庚烷和乙酸乙酯的混合溶液中正庚烷和乙酸乙酯的体积比为:10:0.1~1。
进一步的,步骤3)中获得中间体c的具体方法为:中间体b精品,加入到四氢呋喃中,10℃~30℃下,搅拌溶解形成溶液,降温至0℃~10℃,向其中加入10%naoh水溶液,持续搅拌0~2小时,得黄色澄清溶液,减压蒸馏出四氢呋喃得中间体c。
本发明的有益效果:
通过对每一步中间体、产物的纯化,从而得到符合要求的高纯度终产品,同时生产工艺简单、易于进行产品收集,产品纯度高,每步收率较高的精制纯化方法,为工业生产的进行提供了新的方式,特别是中间体b通过纯化固体,克服了现有技术中以溶液形式进行下一步反应造成杂质较多,后续难以纯化的问题。本发明氢溴酸调节调节溶液ph值,仅收集ph值在6~5阶段萃取的有机相,以达到纯化终产品的目的。
附图说明
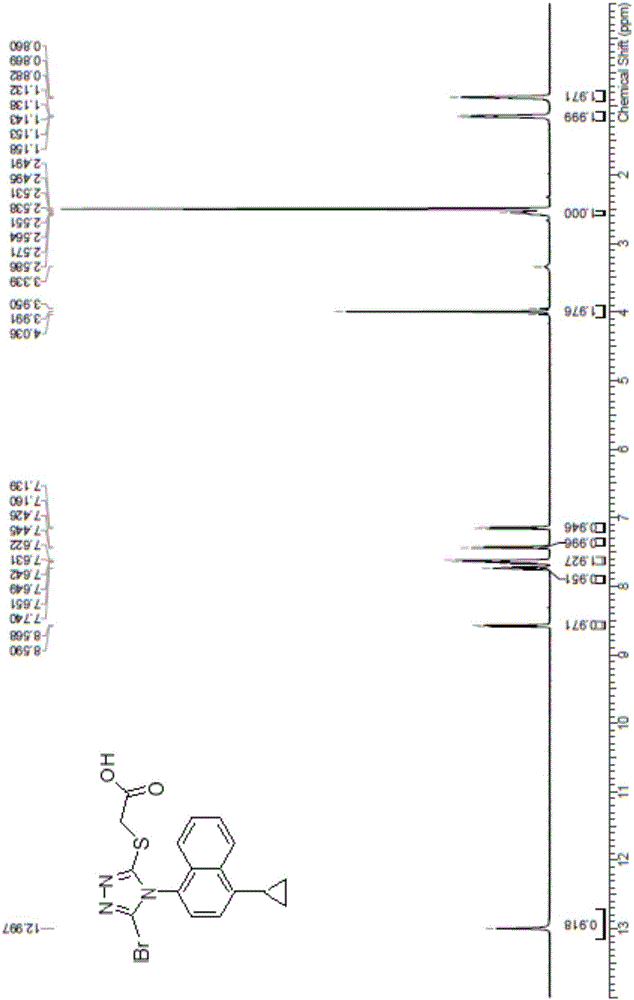
图1为本发明制备的雷西纳德原料药氢谱;
图2为本发明制备的雷西纳德原料药质谱。
具体实施方式
除非特别说明,以下实施例中选用的试剂均为市售的普通试剂,下述实施例中的实验方法,如无特殊说明,均为常规方法。
实施例1
1)向1000ml的三口反应瓶中加入反应溶剂dmf400ml,然后从三颈烧瓶口再加入100g起始物料4-(4-环丙基萘-1-基)-1h-1,2,4-三唑-5(4h)-硫醇,15~20℃下搅拌溶解,获得a溶液。向溶液a中加入粉末状无水碳酸钠39.7g,10~15℃下,滴加入65.7g溴乙酸乙酯,控制滴加速度,滴加过程中内温不超过15℃,滴加完毕后,维持内温10~15℃,保温反应,经薄层色谱,原料反应完全,继续延长反应15分钟后停止反应。
将反应液转移至5l烧杯中,在体系内温0~5℃下,向其中滴加入0~5℃纯化水1.2l,有大量沉淀产生,继续于0~5℃搅拌0.5h,抽滤,至基本无液体流出,沉淀以纯化水(500ml)洗涤2次,抽滤,至基本无液体流出,得白色沉淀。
将沉淀转移至5l烧杯中,30~40℃下向其中加入纯化水0.6l,乙酸乙酯1.2l,开启搅拌,使其完全溶解,继续搅拌10分钟,静置,分液,乙酸乙酯相用纯化水0.5l,洗涤1次,水相用乙酸乙酯0.1l萃取一次,合并有机相加入无水硫酸钠0.15kg,25~30℃下干燥1h。过滤,有机相在35℃下,将部分乙酸乙酯溶液减压旋干得中间体a粗品。
向配有温度计2l三颈烧瓶中,加入未蒸馏乙酸乙酯溶液0.4l,然后从三颈烧瓶口加入已蒸干中间体a粗品,开启搅拌,升温至70~80℃,维持该温度使其完全溶解,缓慢降至10~15℃,有大量沉淀产生,继续于10~15℃搅拌2h,抽滤,用正庚烷:乙酸乙酯=1:1溶剂0.2l洗涤沉淀2次,抽滤,至基本无液体流出,得白色沉淀。
将得到的白色沉淀放入鼓风干燥箱中,40~45℃干燥,得中间体精品a,纯度99.913%,收率90.25%。
2)向配有温度计1l三颈烧瓶中,加入反应溶剂四氢呋喃700ml,开启搅拌,然后从三颈烧瓶口加入100g中间体a。维持内温在25~30℃,搅拌使之完全溶解,得淡黄色溶液。
将溶液内温降至15±2℃,向溶液中分4批加入粉末状n-溴代丁二酰亚胺70.1g,维持体系内温在15±2℃,加料毕,缓慢升温至内温不超过25℃,保温反应,经薄层色谱,原料反应完全,延长反应15分钟后,停止反应。
缓慢降温至0~5℃,向反应液中滴加入3%焦亚硫酸钠水溶液0.3l,继续于0~5℃温度下,向体系中滴加入纯化水至总体积为2l,有大量沉淀产生,保温搅拌0.5h。抽滤,至基本无液体流出,沉淀以四氢呋喃:水=1:4(200ml)洗涤沉淀2次,再以水(l)洗涤沉淀1次,抽滤,至基本无液体流出,得白色沉淀。
将沉淀转移至5l塑料烧杯中,在20~25℃下,向其中加入纯化水1.1l,乙酸乙酯1.1l,开启搅拌,使其完全溶解,向其中加入10%溴化钠水溶液450ml,继续搅拌10分钟,静置,分液,向水相中加入乙酸乙酯450ml,10%溴化钠水溶液200ml,萃取,静置,分液,合并有机相,加入无水硫酸钠150g,干燥1小时,过滤,有机相在35℃下,减压蒸干,得黄色液(固)体。
在15~20℃下,向配有温度计三颈烧瓶中,加入精制溶剂四氢呋喃250ml,开启搅拌,然后从三颈烧瓶口再加入中间体粗品,使其完全溶解,向溶液中加入正庚烷380ml,有大量沉淀产生,缓慢升温至55±5℃,使其完全溶解,在此温度下,继续向溶液中滴加入正庚烷100ml,加料毕,缓慢温至10~15℃,有大量沉淀产生,继续于10~15℃搅拌2小时,抽滤,用正庚烷:四氢呋喃=7:1溶剂(200ml)洗涤沉淀2次,抽滤,至基本无液体流出,得白色沉淀。
将得到的沉淀放入真空干燥箱中,20~30℃干燥,得中间体精品b,纯度99.573%,收率91.26%。
3)在20~30℃水浴中,向配有温度计1l三颈烧瓶中,加入反应溶剂四氢呋喃200ml,开启搅拌,然后从三颈烧瓶口加入100g中间体b。将体系降温至0~5℃,加入10%氢氧化钠100ml,滴加毕,保温反应,体系由白色混悬状态逐渐溶解澄清,经薄层色谱,原料反应完全,延长反应15分钟后,停止反应,得黄色溶液c。
在30~35℃水浴中,将中间体c溶液转移至旋转蒸发仪中,将反应溶剂四氢呋喃蒸出。
4)向中间体c水溶液中补加入纯化水0.3l,然后再加入乙酸乙酯0.2l,搅拌5分钟,静止分液,分去有机相,得黄色水溶液。
在25~30℃下,向其中加入25%氢溴酸溶液,调节水相ph值在6.5~7,向黄色水溶液中加入乙酸乙酯0.2l,搅拌5分钟,静止分液,分去有机相,得黄色水溶液。
在25~30℃下,向其中加入25%氢溴酸溶液,调节水相ph值在5.1~5.5,黄色水溶液中加入乙酸乙酯0.4l,搅拌5分钟,静止分层,检测水相ph值在5.6~6。
在25~30℃下,重复上述步骤,直到检测水相ph值在5.0~5.3之间,合并萃取水相在5~6之间乙酸乙酯相。
雷西纳德精制:将上述乙酸乙酯相转移至蒸馏装置中,开启搅拌,20~25℃下,减压蒸馏,体系由澄清变浑浊,关闭减压,缓慢升温至40±2℃,保温搅拌2小时,降温至20~25℃,继续减压至约3.5~4倍体积(为原料y与乙酸乙酯的质量体积比)关闭减压,缓慢升温至40±2℃,向其中缓慢滴加入正庚烷75ml,加料毕,缓慢降温至0~5℃,继续于0~5℃搅拌2小时,抽滤,用正庚烷:乙酸乙酯=1:1溶剂(90ml)洗涤沉淀2次,抽滤,至基本无液体流出,得白色沉淀。
将得到的沉淀放入真空干燥箱中,40~50℃干燥,得终产品,其氢谱如图1所示,质谱如图2所示,纯度99.898%,收率82.54%。