并五苯及其衍生物的合成方法与流程
本发明属于有机合成领域,具体涉及一种并五苯及其衍生物的合成方法。
背景技术:
并五苯(Pentacene)为五个苯环稠和而成的多环芳香性化合物,并五苯及其衍生物是已被分离和纯化的含苯环数量最多的并苯类碳环化合物系列,并且具有与非晶硅相当的薄膜晶体管迁移率,也是目前加工薄膜晶体管的常用材料。在有机电子领域,并五苯具有成本低易于大面积加工的优势,并因其基于π共轭体系的电化学性能,高的薄膜晶体管的载流子迁移率而被广泛研究。并五苯由于其多芳环分子间的π-π相互作用力,使其不易溶于一般有机溶剂,为改善其溶解性一般需引入取代基团。
近年来,对于过渡金属钯催化的临时导向基团导向的苄基C(sp3)-H直接芳基化反应的研究报道众多,我们首次将其应用于合成并五苯前体化合物。临时导向基团,就是在反应过程中很弱的导向基团与一些配体结合,在原位临时生成一个导向基团,生成的导向基团与过渡金属和导向基团邻位碳氢键形成环过渡金属中间体,活化邻位碳氢键,再通过级联反应实现邻位碳氢键官能团化,最后配体与弱导向基团分离,还原为底物本身结构。因此,该方法是一种快速构建弱导向基团邻位碳氢键官能团化合物骨架的简易方法。
目前,合成含有取代基的并五苯衍生物的方法都有其局限性,如:前体化合物不易获得、工艺繁琐、产率偏低或工艺条件苛刻等,不适合产业化和系列化。
CN 1592729A公开了一种并五苯及其衍生物的制备方法,包括以下步骤,首先由邻苯二甲醛(取代邻苯二甲醛)与1,4-环己酮生成并五苯二醌(取代并五苯二醌);然后将其还原成并五苯二醇(取代并五苯二醇);再将并五苯二醇(取代并五苯二醇)脱水生成6-并五苯醇(取代6-并五苯醇);再将6-并五苯醇(取代6-并五苯醇)还原成6,13,13-三氢-6-并五苯醇(取代6,13,13-三氢-6-并五苯醇);最后将其脱水生成并五苯(并五苯衍生物)。该技术方案提供的一种并五苯及其衍生物的制备方法步骤繁多,不易于实现工业化。
技术实现要素:
本发明针对现有技术的不足,目的在于提供一种并五苯及其衍生物的合成方法。
为实现上述发明目的,本发明采用的技术方案为:
一种并五苯及其衍生物的合成方法,包括如下步骤:
(1)将二碘苯、邻甲基苯甲醛类化合物、醋酸钯(Pd(OAc)2)、三氟醋酸银(AgTFA)、甘氨酸(glycine)按一定摩尔比混合在醋酸(AcOH)和水(H2O)的混合溶剂中,反应一段时间后得到反应液;所述二碘苯为对二碘苯或间二碘苯;
(2)将步骤(1)所得反应液抽滤、洗涤后,再用饱和碳酸氢钠水溶液和二氯甲烷萃取;向萃取液中加入干燥剂,随后过滤,再向滤液中加入硅胶粉,然后用旋转蒸发器旋干二氯甲烷,通过柱层析法分离得到2,2′-(1,4-亚苯基双(亚甲基))二苯甲醛或2,2′-(1,3-亚苯基双(亚甲基))二苯甲醛类化合物;
(3)将步骤(2)所得2,2′-(1,4-亚苯基双(亚甲基))二苯甲醛或2,2′-(1,3-亚苯基双(亚甲基))二苯甲醛类化合物和三氟甲磺酸(TfOH)按一定摩尔比加入到溶剂中,室温下反应一段时间后得到反应液;
(4)将步骤(3)所得反应液用饱和碳酸氢钠水溶液和二氯甲烷萃取;向萃取液中加入干燥剂,随后过滤,向滤液中加入硅胶粉,用旋转蒸发器旋干二氯甲烷,通过柱层析法得到纯净的并五苯及其衍生物。
上述方案中,步骤(1)中所述邻甲基苯甲醛类化合物的分子结构式为:
其中R为甲基、甲氧基、氟、氯和溴中的一种。
上述方案中,步骤(1)所述二碘苯和邻甲基苯甲醛类化合物的摩尔比为1:2.2~2.5。
上述方案中,步骤(1)所述醋酸钯(Pd(OAc)2)与二碘苯的摩尔比为1:10~12.5;所述三氟醋酸银(AgTFA)与二碘苯的摩尔比为2.5~3:1;所述甘氨酸(glycine)与二碘苯的摩尔比为0.6~1:1;所述醋酸(AcOH)和水(H2O)的混合溶剂中醋酸和水的体积比为8~10:1,二碘苯与混合溶剂的比例为1mol:6~8L。
上述方案中,步骤(1)中所述反应的温度为90~110℃,反应时间为24~36小时。
上述方案中,所述饱和碳酸氢钠水溶液和二氯甲烷的体积比为1:1;所述干燥剂为无水硫酸钠。
上述方案中,步骤(3)中所述三氟甲磺酸(TfOH)与2,2′-(1,4-亚苯基双(亚甲基))二苯甲醛或2,2′-(1,3-亚苯基双(亚甲基))二苯甲醛类化合物摩尔比为1:15~20。
上述方案中,步骤(3)所述溶剂为1,1,1,3,3,3-六氟异丙醇(HFIP)。
上述方案中,步骤(3)中所述2,2′-(1,4-亚苯基双(亚甲基))二苯甲醛或2,2′-(1,3-亚苯基双(亚甲基))二苯甲醛类化合物与1,1,1,3,3,3-六氟异丙醇(HFIP)溶剂的比例为1mol:8~10L。
上述方案中,步骤(3)中所述反应的时间为3~5h。
本发明以邻甲基苯甲醛类化合物和对(间)二碘苯类化合物为原料,通过如下反应得到并五苯及其衍生物,具体的反应方程式如下:
其中,R为甲基,甲氧基,氟,氯,溴中的一种。
作为本发明的优选方案,所述邻甲基苯甲醛类化合物为2,4-二甲基苯甲醛,以2,4-二甲基苯甲醛和对二碘苯为原料合成3,10-二甲基并五苯,具体的反应方程式如下:
作为本发明的优选方案,所述邻甲基苯甲醛类化合物为邻甲基甲基苯甲醛,以邻甲基甲基苯甲醛和间二碘苯为原料合成并五苯,具体的反应方程式如下:
本发明的有益效果如下:本发明从简单易得的邻甲基苯甲醛类化合物和对(间)二碘苯类化合物出发,首先通过过渡金属钯催化的临时导向基团导向的苄基C(sp3)-H直接芳基化反应合成并五苯前体,该合成方法简单易行,产率较高;然后使用路易斯酸催化剂,在1,1,1,3,3,3-六氟异丙醇溶剂中发生芳香醛类化合物的苯环化反应,即得并五苯及其衍生物。本发明所述方法步骤少,所涉及原料易得,反应条件温和,易于实现工业化。
附图说明
图1为实施例1所得2,2′-(1,3-亚苯基双(亚甲基))二苯甲醛核磁共振氢谱图。
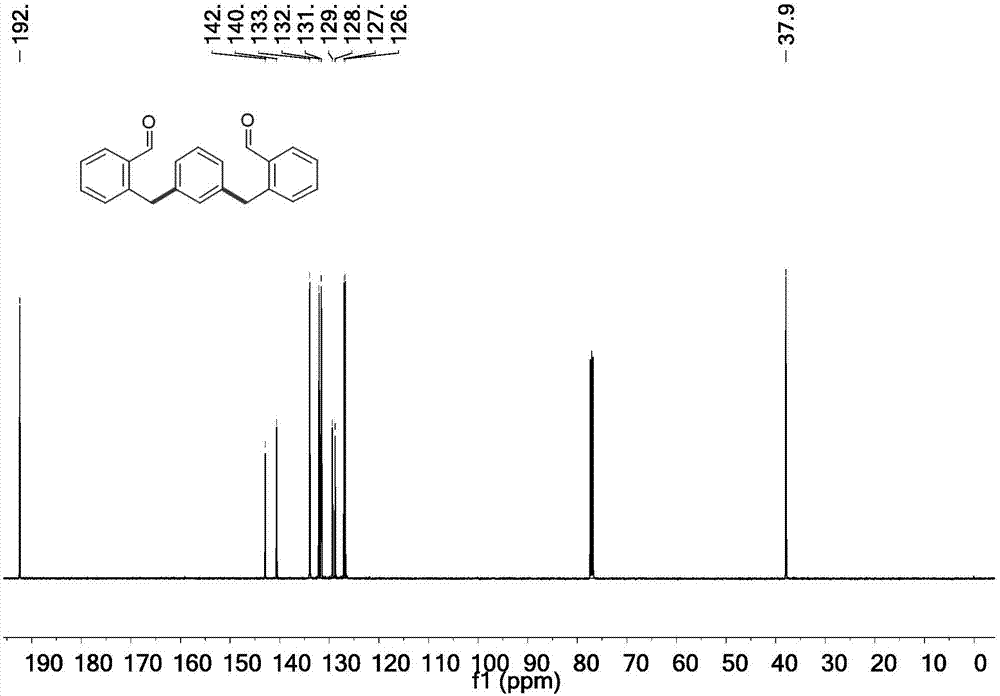
图2为实施例1所得2,2′-(1,3-亚苯基双(亚甲基))二苯甲醛核磁共振碳谱图。
图3为实施例1所得并五苯核磁共振氢谱图。
图4为实施例1所得并五苯核磁共振碳谱图。
具体实施方式
为了更好地理解本发明,下面结合实施例进一步阐明本发明的内容,但本发明的内容不仅仅局限于下面的实施例。
实施例1
将间二碘苯(0.1650g,0.5mmol),邻甲基苯甲醛(0.1502g,1.25mmol),醋酸钯(Pd(OAc)2)(0.0112g,0.05mmol),三氟醋酸银(AgTFA)(0.2761g,1.25mmol),甘氨酸(glycine)(0.0300g,0.4mmol),加入到醋酸(AcOH)和水(H2O)的混合溶剂中,醋酸和水的体积比为9:1,混合溶剂的体积为10mL,在恒温磁力搅拌器中反应,反应温度为100℃,反应时间为24小时。反应结束后,反应液用布氏漏斗抽滤,并用二氯甲烷冲洗2~3次,滤液用饱和碳酸氢钠水溶液和二氯甲烷萃取(80mL:80mL),所得萃取液用干燥剂除水后过滤,向滤液中加入硅胶粉,其中硅胶粉的质量是两种反应物间二碘苯和邻甲基苯甲醛总质量的30~40倍,用旋转蒸发器旋干二氯甲烷,通过柱层析法分离得到2,2′-(1,3-亚苯基双(亚甲基))二苯甲醛(0.1022g,0.33mmol,产率66%,核磁共振氢谱图如图1所示,核磁共振碳谱图如图2所示)。将2,2′-(1,3-亚苯基双(亚甲基))二苯甲醛(0.0629g,0.2mmol),三氟甲磺酸(TfOH)(0.0015g,0.01mmol)加入到6mL 1,1,1,3,3,3-六氟异丙醇(HFIP)中,室温,搅拌,反应5h。反应结束后,用饱和碳酸氢钠水溶液和二氯甲烷萃取(50mL:50mL),所得萃取液用干燥剂除水后过滤,向滤液中加入硅胶粉,其中硅胶粉的质量是反应物2,2′-(1,3-亚苯基双(亚甲基))二苯甲醛质量的30~40倍,用旋转蒸发器旋干二氯甲烷,通过柱层析法分离得到并五苯(0.0462g,0.17mmol,产率83%,核磁共振氢谱图如图3所示,核磁共振碳谱图如图4所示)。
实施例2
将对二碘苯(0.1650g,0.5mmol),2,4-二甲基苯甲醛(0.1677g,1.25mmol),醋酸钯(Pd(OAc)2)(0.0112g,0.05mmol),三氟醋酸银(AgTFA)(0.2761g,1.25mmol),甘氨酸(glycine)(0.0300g,0.4mmol),加入到醋酸(AcOH)和水(H2O)的混合溶剂中,醋酸和水的体积比为9:1,混合溶剂的体积为10mL,在恒温磁力搅拌器中反应,反应温度为100℃,反应时间为24小时。反应结束后,反应液用布氏漏斗抽滤,并用二氯甲烷冲洗2~3次,滤液用饱和碳酸氢钠水溶液和二氯甲烷萃取(80mL:80mL),所得萃取液用干燥剂除水后过滤,向滤液中加入硅胶粉,其中硅胶粉的质量是两种反应物对二碘苯和2,4-二甲基苯甲醛总质量的30~40倍,用旋转蒸发器旋干二氯甲烷,通过柱层析法分离得到2,2′-(1,4-亚苯基双(亚甲基))二(4-甲基苯甲醛)(0.1233g,0.36mmol,产率72%)。将2,2′-(1,4-亚苯基双(亚甲基))二(4-甲基苯甲醛)(0.0685g,0.2mmol),三氟甲磺酸(TfOH)(0.0015g,0.01mmol)加入到6mL1,1,1,3,3,3-六氟异丙醇(HFIP)中,室温,搅拌,反应5h。反应结束后,用饱和碳酸氢钠水溶液和二氯甲烷萃取(50mL:50mL),所得萃取液用干燥剂除水后过滤,向滤液中加入硅胶粉,其中硅胶粉的质量是反应物2,2′-(1,4-亚苯基双(亚甲基))二(4-甲基苯甲醛)质量的30-40倍,用旋转蒸发器旋干二氯甲烷,通过柱层析法分离得到3,10-二甲基并五苯(0.0533g,0.17mmol,产率87%)。
实施例3
将间二碘苯(0.1650g,0.5mmol),2-甲基-4-甲氧基苯甲醛(0.1877g,1.25mmol),醋酸钯(Pd(OAc)2)(0.0112g,0.05mmol),三氟醋酸银(AgTFA)(0.2761g,1.25mmol),甘氨酸(glycine)(0.0300g,0.4mmol),加入到醋酸(AcOH)和水(H2O)的混合溶剂中,醋酸和水的体积比为9:1,混合溶剂的体积为10mL,在恒温磁力搅拌器中反应,反应温度为100℃,反应时间为24小时。反应结束后,反应液用布氏漏斗抽滤,并用二氯甲烷冲洗2~3次,滤液用饱和碳酸氢钠水溶液和二氯甲烷萃取(80mL:80mL),所得萃取液用干燥剂除水后过滤,向滤液中加入硅胶粉,其中硅胶粉的质量是两种反应物间二碘苯和2-甲基-4-甲氧基苯甲醛总质量的30~40倍,用旋转蒸发器旋干二氯甲烷,通过柱层析法分离得到2,2′-(1,3-亚苯基双(亚甲基))二(4-甲氧基苯甲醛)(0.0842g,0.22mmol,产率45%)。将2,2′-(1,3-亚苯基双(亚甲基))二(4-甲氧基苯甲醛)(0.0749g,0.2mmol),三氟甲磺酸(TfOH)(0.0015g,0.01mmol)加入到6mL 1,1,1,3,3,3-六氟异丙醇(HFIP)中,室温,搅拌,反应5h。反应结束后,用饱和碳酸氢钠水溶液和二氯甲烷萃取(50mL:50mL),所得萃取液用干燥剂除水后过滤,向滤液中加入硅胶粉,其中硅胶粉的质量是反应物2,2′-(1,3-亚苯基双(亚甲基))二(4-甲氧基苯甲醛)质量的30-40倍,用旋转蒸发器旋干二氯甲烷,通过柱层析法分离得到2,10-二甲氧基并五苯(0.0602g,0.18mmol,产率89%)。
实施例4
将对二碘苯(0.1650g,0.5mmol),2-氯-4,6-二甲基苯甲醛(0.2108g,1.25mmol),醋酸钯(Pd(OAc)2)(0.0112g,0.05mmol),三氟醋酸银(AgTFA)(0.2761g,1.25mmol),甘氨酸(glycine)(0.0300g,0.4mmol),加入到醋酸(AcOH)和水(H2O)的混合溶剂中,醋酸和水的体积比为9:1,混合溶剂的体积为10mL,在恒温磁力搅拌器中反应,反应温度为100℃,反应时间为24小时。反应结束后,反应液用布氏漏斗抽滤,并用二氯甲烷冲洗2~3次,滤液用饱和碳酸氢钠水溶液和二氯甲烷萃取(80mL:80mL),所得萃取液用干燥剂除水后过滤,向滤液中加入硅胶粉,其中硅胶粉的质量是两种反应物对二碘苯和2-氯-4,6-二甲基苯甲醛总质量的30~40倍,用旋转蒸发器旋干二氯甲烷,通过柱层析法分离得到6,6′-(1,4-亚苯基双(亚甲基))二(2-氯4-甲基苯甲醛)(0.1152g,0.28mmol,产率56%)。将6,6′-(1,4-亚苯基双(亚甲基))二(2-氯4-甲基苯甲醛)(0.0823g,0.2mmol),三氟甲磺酸(TfOH)(0.0015g,0.01mmol)加入到6mL 1,1,1,3,3,3-六氟异丙醇(HFIP)中,室温,搅拌,反应5h。反应结束后,用饱和碳酸氢钠水溶液和二氯甲烷萃取(50mL:50mL),所得萃取液用干燥剂除水后过滤,向滤液中加入硅胶粉,其中硅胶粉的质量是反应物6,6′-(1,4-亚苯基双(亚甲基))二(2-氯4-甲基苯甲醛)质量的30~40倍,用旋转蒸发器旋干二氯甲烷,通过柱层析法分离得到1,12-二氯-3,10-二甲基并五苯(0.0668g,0.18mmol,产率89%)。
显然,上述实施例仅仅是为清楚地说明所作的实例,而并非对实施方式的限制。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而因此所引申的显而易见的变化或变动仍处于本发明创造的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!