可控转录的制作方法
发明背景
干细胞研究为研究人体发育、再生医学、疾病建模、药物研发和细胞移植带来巨大希望。另外,干细胞衍生细胞能够实现研究不轻易可及的人细胞群体的生理和病理反应。这经常涉及研究基因(和其他形式在不编码蛋白质的rna(ncrna)中编码的调节机制)。不幸地,已经证明在人细胞中可控转录或表达遗传信息是特别困难的。
另外,对于再生医学、疾病建模、药物发现和细胞移植的几个关键方面,需要从轻易可及的来源操作和生产成熟的人细胞类型。在人细胞中控制转基因表达是生物学研究的基础。但是,已经证明这在人细胞中困难。另外,真正需求按照适于药物发现和再生医学目的的量和品质,在体外衍生许多高度合乎需要的人细胞类型。因为使干细胞定向分化成所需的细胞类型往往有挑战性,所以其他方案已经浮现,包括将细胞直接再编程成所需的细胞类型。尤其,作为将多能干细胞(包括hpsc)直接转化为成熟细胞类型的方法,正向编程已经被公认为衍生人细胞的强有力策略。这种再编程涉及强迫表达关键谱系转录因子(或非编码性rna,包括lncrna和微rna),旨在将干细胞转化成特定的成熟细胞类型。另外在这种情况下,在人细胞中可控表达遗传信息已经有挑战性。目前可用的正向编程方案主要地基于慢病毒转导细胞法,这导致随机插入的可诱导盒多样化表达或完全沉默。这导致附加的纯化步骤以便分离表达所要求转录因子的子群体。因此,明确需要进一步改进这些方法。
除转基因的诱导型表达之外,迫切需要能够在细胞中控制敲低和敲除基因或其他编码性序列,以允许实施功能丧失研究。在干细胞和成熟细胞类型中的功能丧失研究提供了独特机会以研究调节人类发育、疾病和生理学的机制。但是,现有技术不允许轻易和高效地操作基因表达。将物质如可诱导短发夹rna(shrna)引入干细胞以触发基因敲低的现有技术遭遇随上文讨论的正向再编程见到的许多缺点,如转基因沉默和限制活性的位置效应。因此,需要允许进行干细胞中功能丧失研究的干细胞中诱导型基因敲除和敲低。
对上文方法的任何改进必须确保实现包含于可诱导盒中的遗传物质如转基因的稳定转录,其抵抗沉默和整合位点相关的其他负面影响。沉默可以由多种外遗传机制引起,包括dna甲基化或组蛋白修饰。采用基于慢病毒转导的现有技术方法,获得的细胞是转基因完全表达、部分表达或沉默的不均一群体。显然,这不合乎许多应用的需要。病毒载体展示将其遗传物质整合入基因组转录活跃区域的倾向性,因此增加因插入性诱变所致的致瘤事件的可能性。
对于许多应用,需要控制细胞中所插入遗传物质的转录,从而可诱导盒可以按要求开启并且按特定水平(包括高水平)转录。如果基因组中可诱导盒的插入是随机的,这不能实现。
发明人已经因此开发了一种使得向细胞的基因组稳定引入可诱导盒成为可能,同时能够控制这个可诱导盒转录的方法。在其中需要引入可诱导盒并且控制所插入遗传物质转录的任何细胞类型中、尤其多能干细胞中,这具有益处。可诱导盒可以包含能够转录的任何遗传物质,例如转基因或非编码性rna(ncrna)。将通过从干细胞需要什么作用,包括转基因表达或基因敲低或敲除,确定可诱导盒内部所包含的物质。
发明简述
发明人已经发现,通过使用本文中所述的双重基因组安全港靶向系统,可以插入可诱导盒并控制这种可诱导盒中遗传物质的转录。这种方法是高度合乎需要的,因为所插入遗传物质的外遗传沉默风险降低,并且可以获得转录可诱导盒的均质细胞群体。
本发明因此涉及一种控制细胞中遗传序列转录的方法,所述方法包括以下步骤:
a)将编码转录调节蛋白的基因靶向插入第一遗传安全港位点中;和
b)将可诱导盒靶向插入第二遗传安全港位点中,其中所述可诱导盒包含与诱导型启动子有效连接的所述遗传序列并且所述启动子受转录调节蛋白调节;
其中所述第一和第二遗传安全港位点是不同的。
相对于随机插入基因组,优选可诱导盒特异性整合入基因组安全港位点(gsh)。gsh之前已经定义为“人类基因组的基因内或基因外区域,所述区域能够容纳新整合的dna的可预测表达,而对宿主细胞或生物无不良作用。有用的安全港必须允许插入的遗传序列充分转录,以(通过进一步翻译)产生所需水平的蛋白质或非编码性rna。gsh还不得使细胞有恶变的倾向,也不得改变细胞功能”(sadelain等人,2012,naturereviewscancer,12(1),51–8.doi:10.1038/nrc3179)。
第一遗传安全港位点用来引入编码至少转录调节蛋白的基因。转录调节蛋白(或转录因子)增加基因的基因转录。大部分转录调节物是与有效连接至基因的增强子或启动子近端元件结合的dna结合蛋白。
在一些方面,转录调节蛋白组成型表达并且在细胞中永久表达。转录调节蛋白可以因此与组成型启动子有效连接。组成型启动子在全部生长和发育阶段一致地指导大多数组织和细胞中的基因表达。在本发明方法中使用时,组成型启动子赋予高水平的基因表达。
可以将其他遗传物质(包括基因)连同转录调节蛋白一起插入第一gsh中。这类基因可以包含一个或多个标志物如绿色荧光蛋白(gfp),所述标志物可以用来例如显示已经成功地插入转录调节蛋白。其他选项包括允许基因编辑的基因,例如cas9和衍生物或casl和衍生物,和可以用来分析细胞中特定基因内源或外源表达的报道分子序列。
第二gsh用来引入可诱导盒,在可诱导盒中所需遗传序列与诱导型启动子有效连接。这种启动子仅在受转录调节蛋白正确诱导时才能够实现转录。转录调节蛋白可以受外源供应给细胞的物质控制。因此,外源物质的存在可以允许或阻断来自诱导型启动子的表达。这类可控表达的例子是本文中进一步描述的tet-on系统。
可以将另外的可诱导盒插入其他gsh,所述gsh区别于上文提到的第一和第二gsh。
一个或多个遗传序列可以可控地从第二和/或其他gsh内部转录。实际上,可诱导盒可以含有1、2、3、4、5、6、7、8、9或10个需要插入gsh中并且需要可控诱导其转录的遗传序列。
需要插入gsh或gshs的一个遗传序列或多个遗传序列存在于可诱导盒内部,与诱导型启动子有效连接。这些遗传序列可以是一旦已经诱导启动子的活性,就能够转录成rna的任何合适序列。合适的遗传序列包括但不限于转基因(编码蛋白质的基因,其中产生的rna是翻译成多肽的信使rna(mrna))、非编码性rna(ncrna–包括但不限于shrna、反义rna(asrna)、向导rna(grna)、微rna(mirna)、小干扰性rna(sirna)、反式作用rna(tasirna)、antagomirs、适配体、mirna海绵和任何其他功能性rna)。
可诱导盒可以包含待插入第二或其他gsh中的额外遗传物质。这类额外遗传物质可以包含一个或多个显示转录正在发生的标志物如绿色荧光蛋白(gfp)。备选地或额外地,基因如抗生素或耐药基因可以允许选择成功插入的可诱导盒。另外,可能希望诱导型表达研究其功能的特定基因或将干扰其功能的序列。同等地,可能需要表达增强或阻碍细胞的生物学功能或影响生物的其他部分中的细胞的基因,包括表达生长因子、肽激素,包括胰岛素等。
技术上,向第一和/或第二gsh插入可以发生在一条染色体上或两条染色体上。gsh存在于二倍体生物的两条染色体上的相同基因座处。在两条染色体中插入是有利的,因为它可以使得来自可诱导盒中所插入遗传物质的转录水平增加成为可能,因此实现特别高水平的转录。
可以控制向gsh的插入。可以基于gsh处dna双链断裂(dsb)的定制位点特异性生成,实现向特定gsh中特异性插入遗传物质。随后可以使用任何合适的机制,如同源重组,引入遗传物质。可以使用在基因组中产生特定dsb的任何方法,但是优选的系统包括crispr/cas9和其改变形式、zfns和talen系统。
另外,转录调节物和/或可诱导盒的插入可以设计成可逆的,并且根据需要,可以移除插入的遗传物质和/或替换为备选的转录调节物/可诱导盒。替换转录调节物和/或可诱导盒的方法形成本发明的部分。这类替换可能在这种情况下有用:已经成功地用一种转录调节物和/或一种可诱导盒修饰细胞的培养物,并且需要替换该转录调节物和/或可诱导盒。这利用了已经成功的插入并且可以允许进行较大的插入。为了实施本发明的这个方面,插入可以包括允许从gsh移除全部或部分插入(如一部分插入)的可切割序列。优选的移除或替换方法包括重组方案。
另外,本发明涉及适于向gsh中插入转录调节物和/或可诱导盒的载体。
在一个方面,本发明提供一种控制细胞中转基因表达的方法,所述方法包括以下步骤:
a)将编码转录调节蛋白的基因靶向插入第一遗传安全港位点中;和
b)将与诱导型启动子有效连接的转基因靶向插入第二遗传安全港位点中,其中所述诱导型启动子受转录调节蛋白调节;
其中所述第一和第二遗传安全港位点是不同的。
在本发明的这个方面,先前描述的可诱导盒包含与诱导型启动子有效连接的转基因。在本发明的这个方面,可诱导盒内部所包含的目的遗传序列是转基因,优选地编码蛋白质的基因。因此,可以在细胞内部控制转基因的转录和翻译(表达)。本发明方法的优点是如果需要,它允许转基因过量表达。
另外,在本发明的这个方面,可以将进一步的相同或不同的转基因插入不同于第一和第二gsh的其他gsh。如上文所述,这个转基因与诱导型启动子有效连接。
在一个方面,本发明提供一种控制细胞中非编码性rna转录的方法,所述方法包括以下步骤:
a)将编码转录调节蛋白的基因靶向插入第一遗传安全港位点中;和
b)将可诱导盒靶向插入第二遗传安全港位点中,其中所述可诱导盒包含与诱导型启动子有效连接的编码非编码性rna序列的dna并且所述启动子受转录调节蛋白调节;
其中所述第一和第二遗传安全港位点是不同的。
另外,在本发明的这个方面,可以将进一步的相同或不同的可诱导盒插入不同于第一和第二gsh的其他gsh。这种可诱导盒可以包含与诱导型启动子有效连接的编码非编码性rna序列的dna或任何其他遗传序列并且所述启动子受转录调节蛋白调节。
更具体地,这种方法允许敲低细胞中的内源性基因。因此,本发明提供一种减少细胞中内源性基因转录和/或翻译的方法,所述方法包括以下步骤:
a)将编码转录调节蛋白的基因靶向插入第一遗传安全港位点中;和
b)将可诱导盒靶向插入第二遗传安全港位点中,其中所述可诱导盒包含与诱导型启动子有效连接的编码非编码性rna序列的dna并且所述启动子受转录调节蛋白调节并且其中所述非编码性rna序列阻抑内源性基因转录或翻译;
其中所述第一和第二遗传安全港位点是不同的。
另外,在本发明的这个方面,可以将进一步的相同或不同的可诱导盒插入不同于第一和第二gsh的其他gsh。这种可诱导盒可以包含与诱导型启动子有效连接的编码非编码性rna序列的dna或任何其他遗传序列并且所述启动子受转录调节蛋白调节。
在任何方面或实施方案中,内源性基因可以编码蛋白质或非编码性rna。
在本发明的以上两方面,可诱导盒包含编码非编码性rna(即有功能但不翻译成蛋白质的rna)的dna。这种非编码性rna可以是任何合适的rna,如先前讨论的那些,但优选地是短发夹rna(shrna)。在本发明的稍后方面,非编码性rna可以通过总体上阻断基因转录或翻译或防止表达,以任何合适的方式实现基因敲低。最终,减少或阻断所述基因的表达,但是基因本身保持完好无恙。
备选地,包含于可诱导盒的序列中的非编码性rna可以包括可以用来敲除细胞中内源性基因、尤其用来替换或破坏基因本身的rna。可以用于本发明这个方面的合适的非编码性rna包括crispr/cas9平台的元件,更具体地包括被导引以靶向内源性基因的向导rna(grna)。
因此,在一个方面,本发明提供一种敲除细胞中内源性基因的方法,所述方法包括以下步骤:
a)将编码转录调节蛋白的基因和编码cas9或其衍生物的基因靶向插入第一遗传安全港位点中;和
b)将可诱导盒靶向插入第二遗传安全港位点中,其中所述可诱导盒包含与诱导型启动子有效连接的向导rna并且所述启动子受转录调节蛋白调节,并且其中所述grna序列靶向内源性基因;
其中所述第一和第二遗传安全港位点是不同的。
另外,在本发明的这个方面,可以将进一步的相同或不同的可诱导盒插入不同于第一和第二gsh的其他gsh。这种可诱导盒可以包含与诱导型启动子有效连接的任何遗传序列并且所述启动子受转录调节蛋白调节。
因此,在本发明的上述方面,可控地诱导grna的转录。
在又一个方面,本发明提供一种减少细胞中内源性基因转录和/或翻译的方法,所述方法包括以下步骤:
a)将编码转录调节蛋白的基因靶向插入遗传安全港位点的第一等位基因中;和
b)将可诱导盒靶向插入相同遗传安全港位点的第二等位基因中,其中所述可诱导盒包含与诱导型启动子有效连接的编码非编码性rna序列的dna并且所述启动子受转录调节蛋白调节并且其中所述非编码性rna序列阻抑内源性基因转录或翻译。
另外,本发明提供一种敲除细胞中内源性基因的方法,所述方法包括以下步骤:
a)将编码转录调节蛋白的基因和编码cas9或其衍生物的基因靶向插入遗传安全港位点的第一等位基因中;和
b)将可诱导盒靶向插入相同遗传安全港位点的第二等位基因中,其中所述可诱导盒包含与诱导型启动子有效连接的向导rna并且所述启动子受转录调节蛋白调节,并且其中所述grna序列靶向内源性基因。
这类单步骤敲除或敲低是新颖的并且可以形成本发明的部分。
在一个方面,本发明提供一种将多能干细胞正向编程的方法,所述方法包括步骤:
a)将编码转录调节蛋白的基因靶向插入第一遗传安全港位点中;和
b)将可诱导盒靶向插入第二遗传安全港位点中,其中所述可诱导盒包含与诱导型启动子有效连接的编码关键谱系转录因子的遗传序列,所述诱导型启动子受转录调节蛋白调节;并且
其中所述第一和第二遗传安全港位点是不同的。
可以将另外或附加的可诱导盒插入区别于第一和第二gsh的其他gsh中。
将多能干细胞正向编程为特定成熟细胞类型是高度合乎需要的并且可以使用本发明的双重打靶平台实现。下文描述针对某些细胞类型的特定方法。
在一个方面,本发明提供一种从多能干细胞产生肌细胞的方法,所述方法包括步骤:
a)将编码转录调节蛋白的基因靶向插入第一遗传安全港位点中;和
b)将与诱导型启动子有效连接的myod1基因靶向插入第二遗传安全港位点中,其中所述诱导型启动子受转录调节蛋白调节;其中所述第一和第二遗传安全港位点是不同的,
并且在视黄酸存在下培养所述细胞。
myod1基因是编码生肌分化1蛋白的基因。优选地,视黄酸(ra)是全反式ra。
在另一个方面,本发明提供一种从表达myod1的多能干细胞产生肌细胞的方法,所述方法包括在视黄酸存在下培养所述细胞。
优选地,ra是全反式ra。优选地,细胞过量表达myod1。
在又一个方面,本发明提供一种从多能干细胞产生少突胶质细胞的方法,所述方法包括步骤:
a)将编码转录调节蛋白的基因靶向插入第一遗传安全港位点中;和
b)将与诱导型启动子有效连接的sox10、olig2、nkx2.2和nkx6.2基因之任何组合靶向插入第二遗传安全港位点中,其中所述诱导型启动子受转录调节蛋白调节;其中所述第一和第二遗传安全港位点是不同的,
sox-10、olig2、nkx2.2、nkx6.2基因分别编码转录因子sox-10、olig2、nkx2.2和nkx6.2。
附图简述
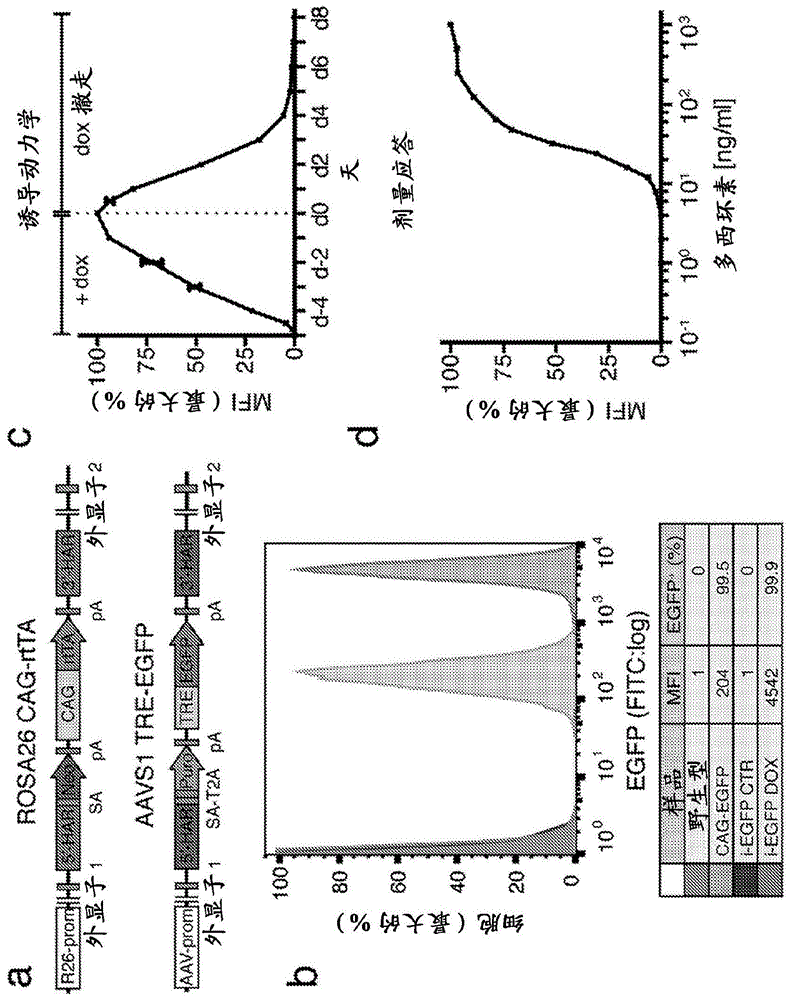
图1(a–d):验证优化的双重基因组安全港靶向过量表达系统。图1(a)hrosa26和aavs1基因座的基因打靶载体的设计。har:同源性臂;sa:剪接受体;t2a:t2a核糖体跳跃信号;neo:新霉素耐药基因;puro:嘌呤霉素耐药基因;pa:多聚腺苷化信号;cag:组成型活性cag启动子;rtta:第三代rtta;tre:诱导型tet应答元件;egfp:增强型绿色荧光蛋白。图1(b)显示表达egfp的hesc中流式细胞术检测的egfp诱导和拯救动力学(1(c))(中位荧光强度,mfi)。结果来自每个时间点的两个生物学重复并且表示为均数±sem。全部值均对5天多西环素后的最大荧光强度(本图中称作第0天)归一化。图1(d)显示用多西环素诱导5天后,表达egfp的hesc中egfp过量表达的多西环素剂量-应答。结果来自每种条件的两个生物学重复并且表示为均数±sem。全部值均对实验中测量的最大荧光强度归一化。用多西环素诱导后,gsh打靶的组成型cag-egfphpsc中和双重gsh打靶的诱导型tre-egfphpsc中的egfp表达水平。纳入野生型hpsc和未诱导的tre-egfp细胞作为阴性对照。
图2(a–d):多西环素(dox)处理后hpsc快速单步骤转化成神经元细胞(i-神经元)的实验方案和结果的概述。图2(a)是这种转化过程的示意图,其中在dox处理后,根据本发明用ngn2转化的细胞诱导分化成神经元细胞。图2(b)显示通过定量rt-pcr-分析证明的i-神经元从hesc生成的正向编程时间过程,其展示泛神经元标志基因(map2、syp)、前脑标志基因(brn2、foxg1)和谷氨酸能神经元标志基因(vglut2,gria4)的时序表达模式。在所示的多西环素处理日分析细胞。相对于内源持家基因pbgd显示各值并且对多能性条件归一化。结果来自每个时间点的三个生物学重复并且表示为均数±sem。图2(c)描述在一周诱导后衍生自hesc的i-神经元中通过免疫染色对βiii-微管蛋白(tubb3)阳性神经元细胞的定量。使用未分化的细胞作为阴性对照(对照),并且报告在新分离的表达ngn2的hesc中和在25代(+p25)后i-神经元生成的数目。图2(d)是借助显示形态变化的系列相差图像描述从hesc生成i-神经元的正向编程时间过程的细胞照片。
图3(a-d):hpsc正向编程为骨骼肌细胞。图3a显示通过诱导型过量表达myod1和视黄酸处理,hpsc快速单步骤转化成骨骼肌细胞的示意图。图3b显示从hesc生成i-肌细胞期间,肌细胞标志基因时序表达模式的定量rt-pcr分析。全部值均相对于hpsc显示。结果来自每个时间点的三个生物学重复并且表示为均数±sem。图3(c)和(d)显示诱导后十天,通过流式细胞术对mhc阳性细胞的定量,表明甚至在培养时间延长和靶向整合myod1系统后传代(p)之后,opti-myod1hpsc保留了它们的生肌效力。使用未分化的细胞作为阴性对照(对照),并且报告在新分离的opti-myod1hesc或在50代(+p50)后的相同细胞中i-肌细胞生成的数字。
图4(a–f):双重gsh打靶的tet-on过量表达系统的打靶策略。图4(a)描述对hpsc中hrosa26和aavs1基因座依次打靶的实验工作流程。关键词:cas9n:来自化脓性链球菌(s.pyogenes)的d10a切口酶突变体cas9核酸内切酶;zfn:锌指核酸酶;neo:新霉素;puro:嘌呤霉素;rtta:第三代逆向四环素反式激活物。该图描述诱导型egfp表达系统(i-egfp)。图4(b)描述hrosa26打靶策略的示意图。图4(c)描述aavs1打靶策略。图4(b)和(c)的关键词:r26-prom:rosa26基因座启动子(thumpd3-as1基因);aav-prom:aavs1基因座启动子(ppp1r12c基因);zfn:锌指核酸酶;5’-har/3’-har:上游/下游同源性臂。sa:剪接受体;t2a:t2a肽;pa:多聚腺苷化信号;cag:cmv早期增强子,鸡β-肌动蛋白和兔β-珠蛋白杂合启动子;tre:tet应答元件;egfp:增强型绿色荧光蛋白。图4(d)描述用来鉴定hrosa26正确打靶和aavs1打靶的hpsc系的基因分型策略示意图;gsh-prom:gsh启动子(分别是hrosa26和aavs1);wt:野生型;可诱导盒:在打靶后整合的整个外源序列。基因座pcr:跨越所靶向基因座的pcr,两条引物均排他性结合于基因组序列外部与同源性臂相对应的基因组dna。注意归因于其高gc含量,cag启动子不能通过常规pcr扩增。因此,正确插入含有cag的表达盒导致pcr扩增子丢失。野生型条带的存在表示存在未打靶的等位基因;野生型条带的丢失表示纯合打靶。5’-int/3’-int:pcr:分别跨越5’-和3’-插入位点的pcr。正确大小的pcr扩增子表示正确整合。3’bbpcr:跨越同源性臂/打靶载体骨架交接处的pcr。pcr产物的存在表示供体质粒的非特异性脱靶整合。图4(e)是凝胶照片,其显示选择的hrosa26-cag-rtta打靶的杂合(het)和纯合(hom)h9hesc的基因分型结果。图4(f)是凝胶照片,其显示选择的aavs1-tre-egfp打靶的杂合(het)和纯合(hom)h9hesc的基因分型结果。1kb+:1kb加dna梯;wt:野生型hesc;pl:打靶质粒;h2o:水对照。
图5(a-e):基于hpsc双重gsh打靶,开发优化的诱导型过量表达平台(opti-ox)。图5(a)显示根据hrosa26基因座和aavs1基因座的一个或两个等位基因是否分别成功地打靶,双重gsh打靶的诱导型egfph9hesc合并成四个实验组。图5(b)显示在成功打靶的杂合和纯合h9hrosa26-cag-rttahesc中,通过蛋白质印迹对rtta蛋白的检测。纳入在随机基因组位置携带第二代rtta的人esc作为对照样品。α-微管蛋白:加样对照。图5(c)描述在图5(a)中描述的多种双重gsh打靶的诱导型egfphesc的代表性例子的流式细胞分析。图5(d)显示在图5(a)中描述的多种双重gsh打靶的诱导型egfphesc中egfp表达的中位荧光强度(mfi)。在对照条件下(无多西环素,ctr)或在多西环素处理(dox)5天后,通过流式细胞术分析细胞。每个数据点代表独立的克隆系。纳入cag-egfphesc和野生型(wt)hesc用于比较。对多西环素处理组(n=4-5,如所示)的统计分析显示,egfp表达水平在双纯合克隆中最高(单因素anova并联合事后dunnet检验;f(2,10)=25.34,p=0.0001;****p<0.0001;**p=0.0026)。选择这个条件用于其他实验。图5(e)显示在图5(a)中描述的多种双重gsh打靶的i-egfphesc中egfp+细胞的百分数。
图6(a-d):在hpsc中和胚层(germlayer)分化期间对opti-ox平台的表征。图6(a)描述在成功靶向的活hpsc中和在多西环素处理五天后它们分化成三个胚层之后egfp水平的流式细胞分析。设定采集设置以包括高水平的诱导的egfp表达(dox)。未诱导的对照群体(ctr)直接紧邻左y-轴存在。图6(b)和图6(c)显示6(a)中流式细胞术图的总结,包括中位荧光强度(mfi)和egfp+细胞百分数。图6(d)显示纯合多能干细胞中和在分化成三个胚层后egfpmrna表达水平的定量rt-pcr结果的条形图。wt:野生型;
图7:人i-神经元的表征。定量rt-pcr结果显示:用多西环素处理时,多能性因子nanog和oct4快速下调。
图8:肌细胞诱导期间的ra信号传导。该图显示,对肌细胞诱导期间六种类视黄醇和类视黄醇受体的qpcr分析表明,在整个i-肌细胞诱导期间rarα、rarβ和全部三种rxr同工型表达,但是rarγ不表达。a是α,b是β并且g是γ。
图9(a)至图9(c):opti-myod1hesc发育成人i-肌细胞的表征。图9(a)显示opti-myod1hpsc至诱导型肌细胞的正向编程时间过程。用每30分钟以nikonbiostationim时间推移系统所获得的自动化相差图像记录形态变化。比例尺:200μm。图9(b)描述qpcr结果,所述结果显示,用多西环素处理处理opti-ngn2hesc时,多能因子nanog和oct4快速下调(左图)。在肌细胞正向编程期间,全部五种主要的人骨骼肌细胞特异性肌细胞重链同工型(由myh基因家族编码)均强烈地上调(右图)。这些同工型包括两种在胚发育和出生后肌肉发育期间表达的同工型(胚同工型myh3;新生儿同工型myh8)和三种通常在成人骨骼肌中表达的同工型[慢颤搐(i型)纤维中的myh7;快颤搐疲乏抵抗性(类型iia)纤维中的myh2和快速疲乏(类型iix)纤维中的myh1]。相反,猫中代表快颤搐、快速疲乏性肌细胞纤维中构成性mhc同工型的myh4在人类中不以显著的量(<1%)表达并且在整个正向编程时间过程期间也不诱导。图9(c)描述诱导的骨骼肌细胞表达类型广泛的常见标志蛋白,包括f-肌动蛋白(通过alexafluor488缀合的毒伞素毒素可视化)、神经细胞黏附分子(ncam)、桥粒蛋白(des)、肌球蛋白重链(myh)、肌联蛋白(ttn),α-辅肌动蛋白(actn2)和肌钙蛋白t(tnnt),但是不表达成肌细胞祖细胞标志物pax3和pax7。全部样品均用成肌蛋白(myog)复染。比例尺:50μm。dapi:核染色。
图10:这三张图描述了用不同浓度的多西环素诱导opti-myod1hpsc后2天,总myod1、内源myod1和myog的qpcr结果。显示用不同浓度的多西环素诱导后48小时的qpcr结果。相对于内源持家基因pbgd对表达进行作图。
图11:tet-on系统的描述。tet-on由两种组件组成:在顶部,描述激活物盒,其中组成型启动子(cp)驱动rtta表达(逆向四环素反式激活物)。rtta是由突变形式的原核性tet阻抑蛋白(tetr)和转录反式激活物结构域vp16(衍生自单纯疱疹病毒)组成的融合蛋白。在底部,描述应答子结构域。它由诱导型启动子(tre,tet应答元件)和目的基因组成。tre是响应于rtta的人工启动子。它由7个系列tet操纵子(teto7)和一个强力最小cmv启动子(mcmv)组成,其本身无活性并且仅在rtta与七个tet操纵子结合时才召集转录装置。需要多西环素(四环素衍生物)以使突变体tetr与tre结合,导致可诱导盒(在这种情况下为egfp)表达。(pa:多聚腺苷化信号)。
图12(a-d)hpsc正向编程为少突胶质细胞。图12(a)描述了使opti-olig2-sox10hpsc快速转化成少突胶质细胞谱系细胞(i-opc和i-ol)的实验方案的示意图。图12(b)显示对每4天连续传代3次和同步brdu脉冲追踪(各自持续4天)后brdu阳性细胞的定量(p=传代次数)。图12(c)显示在i-少突胶质细胞从hpsc生成期间,对编码髓鞘质相关蛋白(cnp、mag、mbp、mog和plp)的基因的时序表达模式的定量rt-pcr分析。在补充pdgfaa和fgf2的少突胶质细胞培养基中诱导opti-olig2-sox10hpsc。在诱导一周后,撤回促分裂原以能够实现终末分化。相对于内源持家基因pbgd显示全部值并且对多能性条件归一化。结果来自每个时间点的2-3个生物学重复并且表示为均数±sem。图12(d)描述在20天诱导后衍生自opti-olig2-sox10hpsc的i-少突胶质细胞中通过免疫染色对cnp阳性细胞和plp阳性细胞的定量。使用未分化的细胞作为阴性对照,并且报告在新分离的opti-ngn2hpsc中或在50代(+p50)后i-少突胶质细胞的数字。
图13是本发明原理的示意图。基本上,该图描述向本发明的核心处两个不同的遗传安全港位点中的插入。一个插入控制第二插入中可诱导盒内部遗传序列的表达。附加的遗传物质可以包含于如所示的多顺反子载体构建体中。另外,可以靶向多于两个遗传安全港位点,从而可以将多个可诱导盒或其他遗传物质置于第一gsh位点中所安置的调节物控制下。
图14(a至f)是结果描述,所示结果显示基于hspc的双重gsh打靶的可诱导敲低系统开发。图14a显示实验方案–h1–h1启动子,to–tet操纵子,tetr–四环素阻抑蛋白。图14b是为获得表达efgp报道转基因的hesc所生成的转基因性等位基因的示意图,其中可以使用诱导型egfpshrna,沉默所述efgp报道转基因。图14c显示在用所示诱导型egfpshrna和tetr组合打靶的hesc中,四环素不存在或存在5天时egfp的表达(std=野生型标准,opt=经密码子优化的)。使用双重打靶的不携带egfpshrna的hesc作为阴性对照。n.s.=p>0.05(不显著),**=p>0.01,***=p>0.001,相对于无tet和无shrna的相同tetr系。图14d是表达std或opttetr的rosa26打靶的hesc中tetr的代表性蛋白质印迹。het=杂合打靶,hom=纯合打靶。具有stdtetr随机整合的hesc作为阳性参考显示,而wth9hesc是阴性对照。tub4a4a是加样对照。加载多种蛋白质数量以促进定量性比较。图14(e):egfpoptikdhesc中通过流式细胞术(mfi)和qpcr(mrna)测量的egfp敲低和拯救动力学。结果来自每个时间点2份独立培养物。图14(f):egfpoptikdhesc中egfp敲低的四环素剂量-应答曲线。报告了半数最大抑制浓度(ic50)。结果来自每个剂量2份独立培养物,并且显示均数。
图15(a、b和c)对rosa26基因座和aavs1基因座作为真正gsh的验证。图15a显示生成gshegfp报道hpsc以检验分化期间gsh表达背后的实验方案。神经元、少突胶质细胞和星形胶质细胞在含有这些细胞谱系的混合物的大批培养物中获得,而全部其他细胞类型均单独地生成。图15b是rosa26和aavs1egfp报道转基因性等位基因的示意图。r26-prom:rosa26基因座启动子;aav-prom:aavs1基因座启动子;5’-har/3’-har:上游/下游同源性臂;sa:剪接受体;t2a:自我剪切性t2a肽;neo:新霉素耐药性;puro:嘌呤霉素耐药性;pa:多聚腺苷化信号;cag:cag启动子;egfp:增强型绿色荧光蛋白。图15(c):在用所示诱导型egfpshrna和tetr组合打靶的hesc中,四环素不存在或存在5天时egfp的表达(野生型标准tetr、stdtetr或密码子优化的tetr、opttetr)。使用双重打靶的不携带egfpshrna的hesc作为阴性对照。结果来自每种条件2-3个独立系(表1)。n.s.=p>0.05(不显著),**=p<0.01,***=p<0.001,相对于无tet和无shrna的相同tetr系。(anova联合事后holm-sidak比较)。
图16(a-d)rosa26和aavs1egfp报道hesc的生成。图16(a):rosa26打靶方案和用来鉴定正确打靶的株系的基因分型策略的示意图。cas9n:来自化脓性链球菌的d10a切口酶突变体cas9核酸内切酶。r26-prom:rosa26基因座启动子(thumpd3-as1基因);5’-har/3’-har:上游/下游同源性臂;转基因:基因打靶后整合的区域;基因座pcr:野生型rosa26基因座的pcr产物(表示未打靶的等位基因);基因座pcr/等位基因丢失:打靶的等位基因的pcr产物/如果转基因含有富含gc的cag启动子则失败的pcr(表示预期的转基因打靶);5’int/3’intpcr:转基因5'末端/3’末端整合区域的pcr产物(表示预期的转基因打靶);5’bb/3’bbpcr:载体骨架5'末端/3’末端的pcr产物(表示非特异性脱靶质粒整合)。注意相似的打靶策略和基因分型策略应用于aavs1基因座打靶。图16(b):为测试组成型egfp(增强型绿色荧光蛋白)表达的最好策略所产生的rosa26转基因性等位基因的示意图。endo-egfp:受内源rosa26启动子驱动的egfp(r26-prom;打靶载体pr26-puro_endo-egfp);ef1α-egfp:受延伸因子1α启动子驱动的egfp(打靶载体pr26-neo_ef1α-egfp);cag-egfp:受cag启动子驱动的egfp(打靶载体pr26-neo_cag-egfp);sa:剪接受体;puro:嘌呤霉素耐药性(嘌呤霉素n-乙酰转移酶);neo:新霉素耐药性(新霉素磷酸转移酶ii);pa:多聚腺苷化信号。图16(c):对代表性rosa26-egfp报道hesc克隆系或野生型h9hesc中egfp阳性细胞(egfp+;显示设门)百分数和egfp中位荧光强度(mfi)的流式细胞定量。图16(d):rosa26-egfp报道hesc中egfp阳性细胞的百分数。结果针对每种条件下进行杂合rosa26打靶的3个克隆。
图17.对hpsc分化后优化的可诱导敲低平台的验证。该图显示在衍生自egfpoptikd(ikd)和soptikd(sikd)hesc的所示细胞类型中四环素不存在(ctr)或存在5天(tet)时,通过qpcr测量的egfp表达。相对于对照条件,对每个独立谱系报道相同系中的egfp水平。缩略语显示图15中描述的谱系(pluri:未分化的)。结果来自每种条件两份独立培养物。
图18(a–d)。在hpsc中开发优化的诱导型crispr/cas9敲除平台。图18a显示用于生成可诱导敲除(iko)hpsc的实验方案。图18b描述了用诱导型grna盒产生aavs1打靶载体的克隆程序的示意图。图18c显示为获得表达egfpd2报道转基因的hesc所生成的转基因性等位基因,其中可以使用诱导型egfpgrna(egfpsoptikohescs),借助crispr/cas9敲除所述报道转基因。bsd:杀稻瘟素耐药性;egfpd2:去稳定化的egfp。图18(d):对来自图19c(grna2–to)和b(grna3–2to)的soptiko细胞中egfpd2可诱导敲除动力学的流式细胞定量。每日在添加四环素后监测egfp阳性细胞百分数。结果来自2份独立培养物。
图19(a至e):在hesc中开发优化的诱导型crispr/cas9敲除平台。(a-d)描述在携带所示grna(2或3)和诱导型启动子组合(to或2to,参见图19e)的egfpd2纯合soptikohesc中egfpd2表达的代表性流式细胞术。打靶载体:paav-puro_sikoegfp-2(19a)、paav-puro_siko-2to-egfp-2(19b)、paav-puro_siko-egfp-3(19c)、paav-puro_siko-2to-egfp-3(19d)。将细胞在四环素(tet)存在下培养5天,或在无四环素的对照(ctr)条件下维持。注意,直方图已经如此归一化,从而对于展示的全部样品,曲线下面积等于1(100%),旨在促进直接目视比较。图19(e):含有一个或两个tet操纵子(分别为h1-to和h1-2to)的soptiko系统的诱导型h1poliii启动子的核苷酸序列。突出显示关键序列特征。显示用于grna克隆的限制性酶切割位点(图18b)。dse:远端序列元件;pse:近端序列元件;teto2:tet操纵子;+1:rna转录起始位置。
图20至图33是在本申请的实施例中所用多种质粒的图的描述。这些质粒是:
20)pspcas9n(bb)_r26-r
21)pspcas9n(bb)_r26-l
22)pr26_cag_egfp
23)pr26_cag_rtta
24)pzfn-aavs1-l-eld(锌指核酸酶(左))
25)pzfn-aavs1-r-kkr(锌指核酸酶(右))
26)paav_cag_egfp(供体)
27)pr26-neo_cag-opttetr(对密码子优化的tetr的hrosa26打靶)
28)paav-puro_ikd(对诱导型shrna的aavs1打靶)
29)paav-neo_cag-cas9(对cas9的aavs1打靶)
30)paav-puro_siko(对诱导型grna的aavs1打靶)
31)paav-puro_siko-2to(对诱导型grna的aavs1打靶,启动子中具有2个tet操纵子的形式)
32)paav_tre-egfp(egfp诱导型过量表达,已连接)
33)paav_tre-myod1(肌肉的myod1诱导型过量表达)
发明详述
发明人开发了一种可用于真核细胞中和尤其多能干细胞及其后代中包含于可诱导盒中的遗传序列诱导型转录的方法。
它特别地适用于通过在所述干细胞内部过量表达促进特定成熟细胞类型发育的可诱导盒,对多能干细胞的正向编程。另外,它还适用于在细胞内部敲低或敲除内源功能,旨在这些细胞中研究功能丧失或改变细胞功能或行为。敲低或敲除可以应用于编码蛋白质的基因或编码非编码性rna的dna序列。可以用本发明方法通过敲除或敲低靶向任一者。
这种方法基于采用在两个或更多个gsh上分摊的诱导型转录的系统,对干细胞基因组中安全港位点的至少双重打靶。但是,这种方法不限于干细胞,并且可以用来例如在研究中或在基因治疗中修饰任何细胞类型的基因组。在本发明的方法中,修饰一个gsh以含有为诱导包含于可诱导盒中的遗传序列转录所需要的转录调节物,其中所述可诱导盒插入基因组中其他地方的不同gsh中。转录调节物优选地组成型表达。优选必须供应外源物质/物质,旨在控制转录调节蛋白的活性并且因此控制可诱导盒的表达。由于本发明方法中使用至少两个独立的gsh,所以存在总计四个可能的插入基因座,因为每个gsh存在于二倍体生物的两条染色体上。如果使用本发明的方法修饰全部四个基因座,则这增加了可能来自细胞的转录量。图5a中显示靶向插入的多种结果的例子。另外,本发明的方法使用至少两个不同的gsh位点。应当理解,其他gsh位点可以用来引入其他转录调节物、可诱导盒或任何其他遗传物质,包括但不限于选择标记、抗生素或耐药基因、与crispr/cas9系统有关的基因或功能未知的基因。
因此,本发明涉及一种控制细胞中所插入遗传序列表达的方法,所述方法包括以下步骤:
a)将编码转录调节蛋白的遗传序列靶向插入第一遗传安全港位点中;和
b)将可诱导盒靶向插入第二遗传安全港位点中,其中所述可诱导盒包含与诱导型启动子有效连接的所述遗传序列,并且所述启动子受转录调节蛋白调节;
其中所述第一和第二遗传安全港位点是不同的。
另外,在本发明的这个方面,可以将进一步的相同或不同的可诱导盒插入不同于第一和第二gsh的其他gsh。这种可诱导盒是如本文所述的。
相对于基因组随机整合,优选在遗传安全港位点内部特异性插入,因为预计这种插入是更安全的基因组修饰,并且更少可能导致不想要的副作用,所述副作用如使天然基因表达沉默或造成导致癌细胞类型的突变。
基因安全港(gsh)位点是基因组内部的基因座,其中可以插入基因或其他遗传物质,同时对细胞或对插入的遗传物质无任何有害作用。最有益的是这样的gsh位点,其中所插入基因序列的表达不受来自相邻基因的任何通读表达扰动并且可诱导盒的表达最大限度减少对内源转录程序的干扰。已经提出更正式的标准,所述标准辅助未来确定特定基因座是否为gsh位点(papapetrou等人,2011,naturebiotechnology,29(1),73–8.doi:10.1038/nbt.1717.)这些标准包括位点(i)与任何基因的5'末端相距50kb或更远,(ii)与任何癌相关基因相距300kb或更远,(iii)与任何微rna(mirna)相距300kb或更远,(iv)位于转录单位之外和(v)位于超级保守区域(ucr)之外。它可以不必要满足提出的全部这些标准,因为已经鉴定的gsh并未满足全部标准。认为一个合适的gsh将满足这些标准的至少2、3、4项或全部。
可以通过寻找其中不破坏天然基因表达的情况下病毒天然整合的位点,鉴定其他位点。
任何合适的gsh位点可以用于本发明的方法中,前提是该位点允许插入遗传物质,同时对细胞无有害作用,并且允许插入的遗传物质转录。本领域技术人员可以使用这种简化标准鉴定合适的gsh,和/或更正式的上述标准。
对于人类基因组,已经鉴定到几个gsh位点,并且这些位点包括aavs1基因座、hrosa26基因座和clybl基因。也已经提出ccr5基因和hprt基因作为可能的gsh,并且进一步研究可以鉴定这些基因中一个或多个作为人类基因组中的gsh。
腺相关病毒整合位点1基因座(aavs1)位于人第19染色体上的蛋白质磷酸酶1调节亚基12c(ppp1r12c)基因内部,所述基因在人组织中一致且普遍地表达。这个位点充当aav血清型2的特定整合基因座,并且因此被鉴定为可能的gsh。aavs1已经证实是有利转录环境,因为它包含开放式染色质结构和能够实现可诱导盒对沉默抵抗的天然染色体绝缘子。不存在因破坏ppp1r12c基因而对细胞所致的已知不良影响。另外,插入这个位点中的可诱导盒在许多不同的细胞类型中保持转录活性。aavs1因此视为gsh并且已经广泛用于人类基因组中靶向转基因。
已经基于与来自小鼠的gsh(rosa26–反向剪接接纳位点#26)的序列相似,鉴定hrosa26位点。尽管已经在人类中鉴定直向同源物位点,但这个位点通常不用于可诱导盒插入。发明人已经开发了专门针对hrosa26位点的靶向系统并且因此能够将遗传物质插入这个基因座中。hrosa26基因座在第3染色体上(3p25.3),并且可以在ensembl数据库内部找到(genbank:cr624523)。该整合位点的精确基因组坐标是3:9396280-9396303:ensembl。该整合位点位于thumpd3非编码性长rna(反向链)的可读框(orf)内部。由于hrosa26位点具有内源启动子,插入的遗传物质可以利用这个内源启动子,或备选地可以与启动子有效连接的情况下插入。
在第13染色体的长臂上的柠檬酸裂合酶β样(clybl)基因的内含子2被鉴定为合适的gsh,因为它是噬菌体衍生phic31整合酶的已鉴定整合热点之一。研究显示,向这个基因座中随机插入的可诱导盒稳定并且表达。已经显示,在这个gsh插入可诱导盒不扰动局部基因表达(cerbibi等人,2015,plosone,doi:10.1371)。clybl因此提供可以适用于本发明中的gsh。
位于第3染色体上(位置3p21.31)的ccr5是编码hiv-1主要共同受体的基因。使用这个位点作为gsh的兴趣源自这个基因中的无效突变,所示无效突变似乎无不良作用,但是预置hiv-1感染抗性。已经开发了靶向第三外显子的锌指核酸酶,因此允许在这个基因座插入遗传物质。鉴于ccr5的天然功能尚待阐明,该位点仍为可能用于本发明的推定性gsh。
次黄嘌呤-鸟嘌呤磷酸核糖转移酶(hprt)基因编码在通过嘌呤补救途径产生嘌呤核苷酸中发挥核心作用的转移酶。因此,需要进一步研究以确保在这个位点处插入不破坏正常细胞功能。但是,已经提出它作为gsh位点。在这个位点处插入可能更适用于成熟细胞类型,如基因治疗的调整。
已经鉴定到其他生物中的gsh并且包括小鼠中的rosa26、hrpt和hipp11(h11)基因座。哺乳动物基因组可能包括基于假attp位点的gsh位点。对于这类位点,已经开发了hic31整合酶(链霉菌属(streptomyces)噬菌体衍生的重组酶)作为非病毒性插入工具,因为它具有将携带attb位点的含有可诱导盒的质粒整合入假attp位点的能力。
gsh还存在于植物的基因组中,并且对植物细胞的修饰可以形成本发明的部分。已经在稻基因组中鉴定到gsh(cantos等人,front.plantsci.,26june2014,volume5,article302,http://dx.doi.org/10.3389/fpls.2014.00302)
在本发明的方法中,在不同的gsh处实施插入,因此本发明的方法需要至少两个gsh。通过插入转录调节蛋白修饰第一gsh。通过插入可诱导盒修饰第二gsh,所述可诱导盒包含与诱导型启动子有效连接的遗传序列。其他遗传物质也可以随这些元件中任一者或两者一起插入。可诱导盒内部与诱导型启动子有效连接的遗传序列优选地是dna序列。可诱导盒的遗传序列优选地编码rna分子,并且因此能够被转录。使用诱导型启动子,控制转录。rna分子可以是任何序列,但优选地是编码蛋白质的mrna、shrna或grna。
第一gsh可以是任何合适的gsh位点。任选地,它是具有组成型表达的内源启动子的gsh;这将导致插入的转录调节蛋白组成型表达。合适的gsh是人细胞的hrosa26位点。备选地,插入的转录调节蛋白与启动子、优选地组成型启动子有效连接。组成型启动子可以与hrosa26位点中的插入联合使用。
转录调节蛋白是这样的蛋白质,其结合于dna、优选地以序列特异性方式结合于位于启动子中或其附近的dna位点,并且促进转录装置与启动子结合,并且因此促进dna序列转录(转录激活蛋白)或阻断这个过程(转录阻抑蛋白)。这类实体也称作转录因子。
转录调节蛋白与之结合的dna序列称作转录因子结合位点或应答元件,并且它们存在于受调节的dna序列的启动子中或其附近。
转录激活蛋白与应答元件结合并促进基因表达。在本发明的方法中优选这类蛋白质以控制可诱导盒表达。
转录阻抑蛋白与应答元件结合并阻止基因表达。
可以通过众多机制激活或失活转录调节蛋白,所述机制包括结合某物质、相互作用于其他转录因子(例如,同型二聚化或异二聚化)或辅调节蛋白、磷酸化和/或甲基化。可以通过激活或失活控制转录调节物。
如果转录调节蛋白是转录激活蛋白,则优选转录激活蛋白需要激活。这种激活可以借助任何合适的手段进行,但优选通过向细胞添加外源物质激活转录调节蛋白。可以控制向细胞供应外源物质,并且因此可以控制转录调节蛋白的激活。备选地,可以供应外源物质以失活转录调节蛋白,并且随后撤回供应以激活转录调节蛋白。
如果转录调节蛋白是转录阻抑蛋白,则优选转录阻抑蛋白需要失活。因此,供应阻止转录阻抑蛋白阻抑转录过程的物质,并且因此允许转录。
可以使用任何合适的转录调节蛋白,优选地使用可激活或可失活的转录调节蛋白。优选可以供应控制转录调节蛋白的外源物质。这类转录调节蛋白也称作诱导型转录调节蛋白。
四环素控制的转录激活是一种诱导型基因表达方法,其中在抗生素四环素或其衍生物之一存在(例如,更稳定的多西环素)下,可逆地开启或关闭转录。在这个系统中,转录激活蛋白是四环素应答的转录激活蛋白(rtta)或其衍生物。rtta蛋白能够与在特定teto操纵基因序列处的dna结合。将这类teto序列的几个重复序列置于最小启动子(如cmv启动子)上游,它们共同形成四环素应答元件(tre)。存在这个系统的两种形式,这取决于添加四环素或衍生物是否激活(tet-on)或失活(tet-off)rtta蛋白。
在tet-off系统中,四环素或其衍生物结合rtta并失活rtta,使它不能够与tre序列结合,因而阻止tre控制的基因转录。这个系统首次在bujard等人(1992).proc.natl.acad.sci.u.s.a.89(12):5547–51中描述。
tet-on系统由两个组件组成;(1)组成型表达的四环素应答的转录激活蛋白(rtta)和rtta敏感的诱导型启动子(tet应答元件,tre)。这个系统可以被四环素或其更稳定的衍生物(包括多西环素(dox))结合,导致激活rtta,从而允许它与tre序列结合并诱导tre控制的基因表达。在本发明的方法中可以优选使用这个系统。图11中描述了这个系统。
因此,转录调节蛋白可以因此是四环素应答的转录激活蛋白(rtta),其可以由外源供应的抗生素四环素或其衍生物之一激活或失活。如果转录调节蛋白是rtta,则插入第二gsh位点中的诱导型启动子包含四环素应答元件(tre)。外源供应的物质是抗生素四环素或其衍生物之一。
可以在本发明方法中使用变体和修饰的rtta蛋白,这些包括tet-on晚期反式激活因子(也称作rtta2s-m2)和tet-on3g(也称作rtta-v16,衍生自rtta2s-s2)。
四环素应答元件(tre)通常由间隔序列分隔的19bp细菌性teto序列的7个重复序列连同最小启动子组成。tre序列的变体和修饰是可能的,因为最小启动子可以是任何合适的启动子。优选地,在rtta结合作用不存在的情况下,最小启动子不显示表达水平或显示最小表达水平。插入第二gsh中的诱导型启动子可以因此包含tre。
基于四环素控制的改进系统是t-rextm系统(thermofisherscientific),其中转录调节蛋白是转录阻抑蛋白tetr。这个系统的组件包括(i)包含强力的人巨细胞病毒立即早期(cmv)启动子和两个四环素操纵基因2(teto2)位点的诱导型启动子,和tet阻抑蛋白(tetr)。teto2序列由2碱基对间隔序列隔开的2个拷贝的19核苷酸序列,5′-tccctatcagtgatagaga-3′组成。在四环素不存在的情况下,tet阻抑蛋白形成同型二聚体,所述同型二聚体以极高亲和力与诱导型启动子中的每个teto2序列结合并阻止自该启动子的转录。四环素一旦添加,则四环素以高亲和力与每个tet阻抑蛋白同型二聚体结合,使其不能与tet操纵基因结合。tet阻抑蛋白:四环素复合物随后从tet操纵基因解离并允许对表达的诱导。在这种情况下,转录调节蛋白是tetr并且诱导型启动子包含两个teto2位点。外源供应的物质是四环素或其衍生物。
本发明还涉及密码子优化的tetr(opttetr)。这可以用于本文所述的任何方法中,或用于其中需要诱导型促进的任何额外用途。使用细菌性tetrcdna序列的多参数优化过程,生成这种实体。与标准序列(stdtetr)相比时,opttetr允许tetr表达增加十倍。在实施例中,tetr的纯合opttetr表达足以防止shrna泄露,同时保留敲低诱导作用。此处包括opttetr的序列,连同作为比较所显示的标准序列。在此要求保护与这个序列具有至少75%、80%、85%或90%同源性、更具体地91%、92%、93%、94%、95%、96%、97%或99%同源性的序列。序列中已经指示在stdtetr和opttetr之间显示待改变的残基,并且优选在opttetr的任何衍生物中不改变这些残基,因为认为这些残基对改善的特性重要。任何衍生物将任选地在所示位置保留这些修饰。
其他诱导型表达系统是已知的并且可以用于本发明的方法中。这些系统包括来自agilenttechnologies的完全控制诱导型系统。这个系统基于可以在哺乳动物细胞中激活转录的昆虫激素蜕皮素或其类似物松甾酮(pona),其中所述的哺乳动物细胞用黑腹果蝇(drosophilamelongaster)蜕皮素受体(ecr)基因和包含蜕皮素受体结合位点的诱导型启动子转染。ecr是核受体的类视黄醇-x-受体(rxr)家族成员。在人类中,ecr与结合于蜕皮素-应答元件(ecre)的rxr形成异二聚体在pona不存在的情况下,转录被该异二聚体阻遏。
因此,转录调节蛋白可以是阻抑蛋白,如蜕皮素受体或其衍生物。后者的例子包括来自agilenttechnologies的vgecr人造受体,其是ecr、糖皮质激素的dna结合结构域和单纯疱疹病毒vp16的转录激活结构域的融合物。该诱导型启动子包含ecre序列或其修饰形式连同最小启动子。修饰的形式包括agilenttechnologies的e/gre识别序列,其中已经对序列做出突变。e/gre识别序列包含类视黄醇-x-受体(rxr)的反转半位点识别元件和gr结合结构域。在全部组合中,外源供应的物质是松甾酮a,其移除ecr或其衍生物对诱导型启动子的阻抑作用,并允许转录发生。
备选地,诱导型系统可以基于合成性类固醇米非司酮作为外源供应的物质。在这种情况下,插入一种杂合转录调节蛋白,其基于酵母gal4蛋白的dna结合结构域、来自人孕酮受体的截短型配体结合结构域(lbd)和来自人nf-κb的激活结构域(ad)。这个杂合转录调节蛋白从thermofisherscientific可获得(geneswitchtm)。米非司酮激活该杂合蛋白,并且允许从包含gal4上游激活序列(uas)和腺病毒e1btata框的诱导型启动子转录。这个系统在wang,y.等人(1994)proc.natl.acad.sci.usa91,8180-8184中描述。
转录调节蛋白因此可以是任何合适的调节蛋白,即,激活蛋白或阻抑蛋白。合适的转录激活蛋白是四环素应答的转录激活蛋白(rtta)或基因开关杂合转录调节蛋白。合适的阻抑蛋白包括rtta、tetr或ecr的tet-off形式。如需要,可以修饰或衍生化转录调节蛋白。
诱导型启动子可以包含适于结合转录调节蛋白或与之相互作用的元件。转录调节蛋白与诱导型启动子的相互作用优选地受外源供应的物质控制。
外源供应的物质可以是与转录调节蛋白结合或相互作用的任何合适物质。合适的物质包括四环素、松甾酮a和米非司酮。
因此,将编码转录调节蛋白的基因插入第一gsh提供了表达可诱导盒的控制机制,所述可诱导盒与诱导型启动子有效连接并插入第二个不同的gsh位点中。
可以提供转录调节蛋白基因以便随其他遗传物质插入。这种物质包括标志物的基因或报道分子的基因,如引起视觉可辨认特征的基因,包括荧光蛋白和发光蛋白。例子包括编码水母绿色荧光蛋白(gfp)的基因(其造成表达它的细胞在蓝色/紫外线下发出绿色光)、催化与萤光素的反应以产生光的萤光素酶和来自基因dsred的红色荧光蛋白。这类标志物或报道基因是有用的,因为报道蛋白的存在确认蛋白质从第一gsh表达,提示插入成功。选择标记可以进一步包括针对抗生素或其他药物的抗性基因。也可以引入能够实现研究内源(或外源)基因表达的标志物或报道基因序列。这包括cas蛋白,所述cas蛋白包括casl、能够实现切除目的基因的cas9蛋白,以及例如通过充当转录增强子或阻抑蛋白来介导其他基因的表达发生变化的cas融合蛋白。另外,可能需要非诱导型表达的分子工具,包括光遗传学工具、核受体融合蛋白,如他莫昔芬诱导型系统ert,和设计者药物独家激活的设计者受体。另外,编码信号传导因子的序列可以从相同的gsh作为转录调节蛋白共表达,其中所述的信号传导因子改变生物中相同细胞或相邻或甚至远距离细胞的功能,包括激素、自分泌因子或旁分泌因子。
另外,如本文中讨论,其他遗传物质可以包括编码非编码性rna的序列。这类遗传物质的例子包括mirna的基因,其可以作为遗传开关发挥作用。
优选编码转录调节蛋白的基因与组成型启动子有效连接。备选地,可以如此选择第一gsh,从而它已经具有也可以驱动转录调节蛋白基因和任何相关遗传物质表达的组成型启动子。组成型启动子确保持久和高水平的基因表达。常用的组成型启动子包括人β-肌动蛋白启动子(actb)、巨细胞病毒(cmv)、延伸因子-1α(ef1α)、磷酸甘油酸激酶(pgk)和遍在蛋白c(ubc)。cag启动子是强力的人造启动子,其通常用来驱动高水平基因表达并且由以下序列构成:(c)巨细胞病毒(cmv)早期增强子元件、(a)鸡β-肌动蛋白基因的启动子、第一外显子和第一内含子,和(g)兔β-珠蛋白基因的剪接受体。
另外,可以向转录调节物连同任何其他遗传物质一起提供可切割序列。这类序列是序列由能够特异性切割dna的实体识别,并且包含限制性位点作为限制性酶的靶序列或其他dna切割实体(如核酸酶、重组酶、核酶或人工构建体)识别的序列。可以包括至少一个可切割序列,但优选地存在两个或更多个可切割序列。这些可切割序列可以在插入物中的任何合适点处,从而可以从gsh选择性地移除选定的插入物部分或全部插入物。该方法因此可以扩展至从gsh移除和/或替换插入物或其部分。可切割位点可以因此在可能需要移除的插入物的部分/全部旁侧分布。使用这种方法,可以移除转录调节物和/或其他遗传物质。
插入物的一部分可以是插入物直至99%–即1-99%、90%、80%、70%、60%、50%、40%、30%、20%、10%或少于10%的任何部分。
可以优选旁侧分布有可切割位点的插入物部分包括组成型启动子。备选地,旁侧分布有可切割位点的部分中不包括组成型启动子。
优选的可切割序列是cre重组酶的loxp位点,因为它允许直接替换移除的插入物。备选地或额外地,可切割序列是dre重组酶的rox位点。
优选在第一gsh处的插入在基因组中的两个基因座发生,因此通过插入修饰每种等位基因。这允许来自编码转录调节物和任何相关遗传物质的基因的表达更大。
第二gsh可以是任何合适的gsh位点。可以优选第二gsh位点不与内源启动子接合,从而插入的可诱导盒的表达仅处于转录调节蛋白的控制下。
可诱导盒包含应转移入细胞的目的遗传序列,优选地dna序列。向基因组中引入可诱导盒具有通过添加允许基因表达或敲低/敲除内源表达的遗传序列来改变该细胞表型的可能性。本发明的方法在细胞中提供了可诱导盒中的遗传序列的可控转录。
用于插入的目的遗传序列优选地是编码rna分子的dna序列。该rna分子可以是任何序列,但优选地是编码性或非编码性rna。编码性或信使rna编码多肽序列,并且这类rna的转录导致蛋白质在细胞内部表达。非编码性rna可以有功能并且可以包括而不限于:微rna、小干扰性rna、piwi相互作用rna、反义rna、小的核rna、小的核仁rna、小cajalbodyrna、yrna、增强子rna、向导rna、核酶、小发夹rna、小时序rna、反式作用rna、小干扰性rna和亚基因组信使rna。非编码性rna也可以称作功能性rna。几个类型的rna在本质上有调节性,并且例如可以通过与mrna或基因的dna的部分互补,下调基因表达。微rna(mirna;21-22个核苷酸)存在于真核生物中并且通过rna干扰(rnai)发挥作用,其中mirna和酶的效应子复合体可以切割互补性mrna、阻断mrna翻译或加速其降解。另一个类型的rna即小干扰性rna(sirna;20-25个核苷酸)通过rna干扰以类似于mirna的方式发挥作用。某些mirna和sirna可以造成它们靶向的基因甲基化,因而减少或增加这些基因的转录。动物具有piwi相互作用rna(pirna;29-30个核苷酸),其在种系细胞中有活性并且认为其是针对转座子的防御。许多原核生物具有crisprrna,一种类似于rna干扰的调节系统,并且这种系统包含向导rna(grna)。反义rna是广泛存在的;大部分下调基因,但是一些是转录的激活物。反义rna可以通过与mrna结合,形成经酶促降解的双链rna来发挥作用。存在许多在真核生物中调节基因的非编码性长rna,这样一种rna是xist,其覆盖雌性哺乳动物中的一条x染色体并且使之失活。因此,存在多种可以用于本发明方法中的功能性rna。
因此,可诱导盒可以包含蛋白质编码基因的遗传序列。这种基因可能不天然地存在于细胞中,或可能天然地存在于细胞中,但是需要该基因的可控表达。备选地,可诱导盒可以是突变、修饰或正确形式的存在于细胞中的基因,尤其用于基因治疗目的或导出疾病模型。可诱导盒可以因此包含来自相同物种的不同生物的转基因(即来自人类的患病/突变形式的基因,或来自人类的野生型基因)或来自不同物种。
在任何方面或实施方案中,包含于可诱导盒中的遗传序列可以是合成性序列。
可诱导盒可以包含需要插入细胞的基因组中的任何合适的遗传序列。因此,遗传序列可以是编码蛋白质产物的基因或这样的序列,所述序列转录成具有功能的核糖核酸(rna)(如核小rna(snrna)、反义rna、微rna(mirna)、小干扰性rna(sirna)、转运rna(trna)和其他非编码性rna(ncrna),包括crispr-rna(crrna)和向导rna(grna)。
可诱导盒可以因此包含需要在细胞中控制其转录的任何遗传序列。选择的遗传序列将取决于细胞类型和修饰后细胞将投向的用途,如下文进一步讨论。
例如,对于基因治疗方法,可能想要以提供野生型基因序列作为可诱导盒的组件。在这种情况下,遗传序列可以是任何编码人类或动物蛋白质的基因。蛋白质编码基因的例子包括人β-珠蛋白基因、人脂蛋白脂肪酶(lpl)基因、人类中由chm基因编码的rab护送蛋白1和更多其他者。备选地,可诱导盒可以表达生长因子,包括bdnf、gdf、ngf、igf、fgf和/或可以切割前肽以形成活性形式的酶。也可以通过表达可诱导盒实现基因治疗,所述可诱导盒包含了编码反义rna、mirna、sirna或在细胞内部干扰另一个基因表达的任何类型rna的遗传序列。
备选地,若细胞为干细胞,则可诱导盒可以包含编码关键谱系特异性主调节物(此处缩写为主调节物)的遗传序列。主调节物可以是以下一者或多者:转录因子、转录调节物、细胞因子受体或信号传导分子等。主调节物是表达的基因,所述基因影响表达它的细胞的谱系。有可能对于待确定的细胞谱系,需要主调节物的网络。如本文所用,通过直接调节或借助基因表达变化级联调节多个下游基因,在发育性谱系或细胞类型的开端表达的主调节物基因参与该谱系的特化。如果主调节物表达,则它有能力重新规定被指定形成其他谱系的细胞的命运。主调节物的例子包括生肌转录因子myod和造血转录因子scl。特别地,主调节物包括但不限于:
神经谱系:少突胶质细胞:sox10、olig2、nkx2.2.、nkx6.2;星形胶质细胞:nfia、nfib和sox9;神经元:ascl1、神经原质蛋白(neurogenin)和neurod、pax6、neurog2、ascl1、dlx2和neurod1;造血细胞,包括红细胞和巨核细胞:gata1、fli1和tal1
间充质谱系:骨骼肌:myod;心肌细胞:gata4、mef2c、baf60c和tbx5;骨:l-myc(rxol)、runx2、osterix、oct4;软骨:c-mycklf4、sox9;和棕色脂肪细胞:c/ebp-β和c-myc
内胚层
胰细胞类型:pdx1和gata6。
干细胞:外胚层sc:oct4、sox2、klf4和c-myc
备选地或额外地,遗传序列或其他遗传物质可以是这样的基因,其中需要研究其功能,从而可控表达可以观察表达对细胞的影响;基因可以包括生长因子和/或细胞因子,以便在细胞移植中使用细胞;和/或基因可以是报道测定法的组分。
另外,遗传序列可以编码其功能是敲低内源性基因表达的非编码性rna或在细胞中编码非编码性rna的dna序列。备选地,遗传序列可以编码crispr-cas9系统的向导rna以实现内源性基因敲除。
本发明的方法因此扩展至敲低细胞内部内源性基因表达的方法。这些方法如前述,并且可诱导盒包含了编码与诱导型启动子有效连接的非编码性rna的遗传序列,其中非编码性rna阻抑所述内源性基因的表达。非编码性rna可以通过任何合适的手段,包括rna干扰和反义rna,抑制基因表达。因此,遗传序列可以编码可以干扰内源性基因的信使rna的shrna。
内源性基因表达减少可以是部分的或完全的–即与诱导非编码性rna转录之前的细胞相比,表达可以减少50%、55%、65%、70%、75%、80%、85%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或100%。
本发明的方法还扩展至通过cripsr-cas9系统,敲除细胞内部内源基因的方法,不过可以使用任何其他合适的基因敲除系统。在这种情况下,优选的是cas9基因组成型表达,并且因此连同转录调节物的基因一并纳入第一gsh中。编码grna的遗传序列可以包含于可诱导盒中,所述可诱导盒插入第二gsh中。grna是短的合成性rna,其由cas9结合作用必需的支架序列和大约20个核苷酸的靶向序列组成,所述靶向序列限定待修饰的基因组靶。因此,可以通过单纯改变grna中存在的靶向序列,改变cas9的基因组靶。尽管这种系统的主要用途是设计靶向内源性基因以敲除基因的grna,也可以修饰它以选择性激活或阻抑靶基因、纯化dna的特定区域和甚至进行dna成像。构思了全部可能的用途。
可诱导盒包含与诱导型启动子有效连接的遗传序列。“启动子”是启动并调节多核苷酸转录的核苷酸序列。“诱导型启动子”是这样的核苷酸序列,其中与启动子有效连接的遗传序列的表达受分析物、辅因子、调节蛋白等控制。在本发明情况下,通过转录调节蛋白实现控制。术语“启动子”或“控制元件”意图包括全长启动子区和这些区域的功能性(例如,控制转录或翻译)区段。“有效连接”指各元件的布局,其中如此描述的组分如此配置,从而履行它们的常规功能。因此,当存在正确的酶时,与遗传序列有效连接的给定启动子能够实现序列的表达。启动子不必要与序列邻接,只要它发挥作用指导所述序列表达即可。因此,例如,不翻译但被转录的间插序列可以存在于启动子序列和遗传序列之间,并且该启动子序列仍可以视为“与遗传序列有效连接”。因此,术语“有效连接”意在涵盖可诱导盒中启动子元件和遗传序列的任何间距或取向,所述间距或取向允许当转录复合体识别启动子元件时可诱导盒的转录起始。
另外,其他遗传物质也可以与诱导型启动子有效连接。其他遗传物质可以包括基因、编码性序列rna、遗传物质,如标志物或报道基因。前文已经讨论这类额外遗传物质。在一些情景下,如若遗传序列本身不是自杀基因,则可能想要在可诱导盒中包含自杀基因用于基因治疗癌症。自杀基因可以使用可诱导盒中的相同诱导型启动子,或它可以拥有独立的诱导型启动子以允许独立控制。这种基因可以用于基因治疗场景,其中如果满足某些条件,则希望能够摧毁供体/转染的细胞。自杀基因是表达造成细胞发生凋亡的蛋白质的基因,或备选地可能需要外部供应的辅因子或辅助药物以起效。辅因子或辅助药物可以由自杀基因的产物转化成高度细胞毒性的实体。
另外,可诱导盒可以包含可切割序列。这类序列是由能够特异性切割dna的实体识别的序列,并且包含限制性位点作为限制性酶的靶序列或其他dna切割实体(如核酸酶、重组酶、核酶或人工构建体)识别的序列。可以包括至少一个可切割序列,但优选地存在两个或更多个可切割序列。这些可切割序列可以在盒中的任何合适点处,从而可以从gsh选择性地移除选定的盒部分或完整盒。该方法因此可以扩展至从gsh移除和/或替换盒或其部分。可切割位点可以因此在可能需要移除的遗传序列的部分/全部旁侧分布。该方法可以导致移除可诱导盒和/或其他遗传物质。
该盒的一部分可以是该盒直至99%–即1-99%、90%、80%、70%、60%、50%、40%、30%、20%、10%或少于10%的任何部分。
可以优选旁侧分布有可切割位点的插入物部分包含与遗传序列有效连接的启动子。备选地,旁侧分布有可切割位点的部分中不包含与遗传序列有效连接的启动子。
优选的可切割序列是cre重组酶的loxp位点,因为它允许直接替换移除的插入物。备选地或额外地,可切割位点可以是dre重组酶的rox位点。
将转录调节蛋白和可诱导盒,连同任何相关遗传物质一起插入细胞的基因组内部不同的gsh中。
向gsh中的插入优选地如前述那样特异性插入gsh序列中。可以使用向特定序列中插入多核苷酸的任何合适技术,并且本领域描述了几种技术。合适的技术包括在目的位置引入断裂并允许载体重组至空位中的任何方法。因此,位点特异性靶向基因组修饰的决定性第一步骤是在待修饰的基因组座位生成双链dna断裂(dsb)。不同的细胞修复机制可以开发用来修复dsb并引入所需的序列,并且这些机制是非同源末端接合修复(nhej),其更易出错;和由供体dna模板介导的同源重组修复(hr),其可以用来插入可诱导盒。
存在几项允许在基因组中位点特异性定制产生dsb的技术。这些技术中许多涉及使用定制的核酸内切酶,如锌指核酸酶(zfn)、转录激活蛋白样效应核酸酶(talen)或成簇规律间隔的短回文重复序列/crispr相关蛋白(crispr/cas9)系统(gaj,t等人“zfn,talenandcrispr/cas-basedmethodsforgenomeengineering,”trendsbiotechnol,31:397–405,2013年7月)。
锌指核酸酶是通过锌指dna结合结构域与限制性酶foki的核酸酶结构域融合所产生的人工酶。后者具有必须二聚化以便切割dna的非特异性切割结构域。这意味着需要两个zfn单体以允许foki结构域发生二聚化及切割dna。dna结合结构域可以设计成靶向任何目的基因组序列,是一系列串联的cys2his2锌指,各自识别靶序列中的三个连续核苷酸。两个结合位点由5-7bp分隔以允许foki结构域的最佳二聚化。该酶因此能够在特定位点切割dna,并且通过确保两个近端dna结合事件必须发生以实现双链断裂,增加靶特异性。
转录激活蛋白样效应核酸酶或talen是二聚体转录因子/核酸酶。通过tal效应子dna结合结构域融合于(核酸酶)dna切割结构域,产生它们。转录激活蛋白样效应(tale)可以经工程化以实际上结合任何所需的dna序列,从而与核酸酶组合时,可以在特定位置切割dna。tal效应子是黄单胞菌属(xanthomonas)细菌分泌的蛋白质,其dna结合结构域含有重复的高度保守的33-34氨基酸序列,同时第12和第13氨基酸趋异。这两个位置高度可变并且显示与特异性核苷酸识别的强相关性。氨基酸序列和dna识别作用之间的这种直白关系允许通过选择在这两个可变位置含有适宜残基的重复区段的组合,工程化特定的dna结合结构域。talen因此从各自靶向单一核苷酸的33至35氨基酸模块阵列建立。通过选择模块阵列,可以靶向几乎任何序列。再次,使用的核酸酶可以是foki或其衍生物。
已经鉴定三个类型的crispr机制,其中最充分地研究了ii型。crispr/cas9系统(ii型)利用cas9核酸酶在dna中由短向导rna决定的位点处产生双链断裂。crispr/cas系统是对外来遗传元件赋予抵抗性的原核免疫系统。crispr是含有碱基序列短重复的原核dna区段。每个重复区之后是来自先前暴露于外来遗传元件的“前间隔区dna(protospacerdna)”的短区段。crispr间隔区识别并使用rna干扰切割外源性遗传元件。crispr免疫应答通过两个步骤进行:crispr-rna(crrna)生物生成和crrna导引的干扰。crrna分子由转录自前间隔区dna的可变序列和crisp重复序列组成。每种crrna分子随后与称作反式激活性crisprrna(tracrrna)的第二rna杂交,并且这两者一起最终与核酸酶cas9形成复合体。如果互补性靶dna序列毗邻于称作前间隔区毗邻基序(pam)的短序列的话,则前间隔区dna编码的crrna部分指导cas9切割互补性靶dna序列。这个天然系统已经工程化并开发用来在基因组dna中特定位点处引入dsb断裂,连同许多其他应用。特别地,可以使用来自化脓性链球菌(streptococcuspyogenes)的cripsrii型系统。在其最简单情况下,crispr/cas9系统包含两个递送至细胞以提供基因组编辑的组件:cas9核酸酶本身和小向导rna(grna)。grna是定制的位点特异性crrna(被指引至靶序列)和标准化tracrrna的融合物。
一旦已经产生dsb,则供应对靶向的基因座具有同源性的供体模板;可以通过同源性指导的修复(hdr)途径修复dsb,允许产生精确插入。
这个系统的衍生物也是可能的。可获得cas9的突变体形式,如cas9d10a,其仅具有切口酶活性。这意味着它仅切割一条dna链并且不激活nhej。相反,当提供有同源性修复模板时,仅通过高保真hdr途径实施dna修复。cas9d10a(congl.等人(2013)science,339,819–823)可以用于配对的cas9复合体中,所述复合体设计成生成毗邻dna切口连同与靶位点对侧链上毗邻区域互补的二个sgrna,这可能是特别有利的。
可以在一种或多种载体(如用于细胞中表达的质粒)中引入用于产生双链dna断裂的元件。
因此,在基因组中产生特异性靶向双链断裂以实现插入基因/可诱导盒的任何方法可以用于本发明的方法中。可以优选用于插入基因/可诱导盒的方法利用zfn、talens和/或crispr/cas9系统或其任何衍生物中的任一者或多者。
一旦已经通过任何适宜的手段产生dsb,则可以按照如下文描述的任何合适方式供应用于插入的基因/可诱导盒。基因/可诱导盒和相关遗传物质形成供体dna以修复dsb处的dna并且使用标准的细胞修复装置/途径插入它们。如上文所示,如何引发断裂将改变哪个途径用来修复损伤。
转录调节蛋白和可诱导盒可以在独立的载体上供应于本发明的方法。“载体”是作为载具用于人工携带遗传物质进入细胞的核酸分子,如dna分子。载体通常是由插入物(如可诱导盒或转录调节蛋白的基因)和充当载体“骨架”的较大序列组成的核酸序列。载体可以处于任何合适的样式,包括质粒、微环或线性dna。载体至少包含与诱导型启动子有效连接的转录调节物的基因或可诱导盒,连同能够实现基因插入有关gsh中的最小序列。任选地,载体还拥有允许载体(例如在细菌中)扩增的复制起点(ori)。额外地或备选地,载体包括选择标记如抗生素耐药基因、有色标记的基因和自杀基因。
图20至图33中描述了实施例中所用载体的例子。
在本发明方法中所用的细胞可以是任何人类细胞或动物细胞。它优选地是哺乳动物细胞,如来自以下者的细胞:啮齿类如小鼠和大鼠;有袋类如袋鼠和树袋熊;非人灵长类如倭黑猩猩、黑猩猩、狐猴、长臂猿和猿;驼类(camelid)如骆驼和羊驼;家畜动物如马、猪、牛、水牛、野牛、山羊、羊、鹿、驯鹿、驴、爪哇野牛、牦牛、鸡、鸭和火鸡;家养动物如猫、犬、兔和豚鼠。细胞优选地是人类细胞。在某些方面,细胞优选地是来自家畜动物的细胞。
本发明方法中所用细胞的类型取决于遗传物质插入gsh位点完成时细胞的用途。
在目的是从祖细胞产生成熟细胞类型的情况下,修饰的细胞是干细胞,优选地是多能干细胞。多能干细胞具有分化成身体内几乎任何细胞的潜力。存在几种多能干细胞源。胚胎干细胞(es细胞)是从胚泡(早期着床前胚胎)的内部细胞团衍生的多能干细胞。诱导型多能干细胞(ipsc)是已经通过被强制表达对维持胚胎干细胞的限定性特性重要的基因和因子,被基因再编程达到胚胎干细胞样状态的成年细胞。在2006年,显示引入四个编码转录因子的特定基因可能使成年细胞转化成多能干细胞(takahashi,k;yamanaka,s(2006),cell126(4):663–76),但是后续工作已经减少/改变了所需基因的数目。oct-3/4和某些sox基因家族成员已经被鉴定为涉及诱导过程的潜在决定性转录调节物。附加基因,包括某些klf家族成员、myc家族、nanog和lin28,可以增加诱导效率。可以收纳于再编程因子中的基因的例子包括oct3/4、sox2、sox1、sox3、sox15、sox17、klf4、klf2、c-myc、n-myc、l-myc、nanog、lin28、fbx15、eras、ecat15-2、tcl1、β-联蛋白(β-catenin)、lin28b、sall1、sall4、esrrb、nr5a2、tbx3和glis1,并且这些再编程因子可以单独使用或与其两者或更多者组合使用。
在目的是产生具有基因敲低或敲除的干细胞供进一步研究的情况下,修饰的细胞可以是干细胞、优选地是多能干细胞,或成熟细胞类型。上文讨论了多能干细胞源。
如果通过插入可诱导盒修饰的细胞待用于人类患者中,可以优选该细胞是衍生自该个体的ipsc。自体细胞的这种用途将消除细胞与接受者匹配的必要。备选地,可以使用市售ipsc,如从
在某些实施方案中,可以优选的是,所用细胞是胚胎干细胞或干细胞系。现在可获得众多胚胎干细胞系,例如,wa01(h1)和wa09(h9)可以从wicell获得,并且khes-1、khes-2和khes-3可以从京都大学再生医科学研究所(京都,日本)获得。
可以优选的是,在不破坏胚胎的情况下衍生胚胎干细胞,特别地在细胞属于人类的情况下,因为这类技术轻易可获得(chung,young等人,cellstemcell,第2卷,第2期,113-117)。还可获得在不破坏胚胎的情况下已经衍生的干细胞系。在一个方面,本发明不扩展至涉及破坏人胚胎的任何方法。
本发明的一个优选方面是将多能干细胞正向编程为成熟细胞类型。因此,本发明的方法可以用于从多能干细胞生产成熟细胞类型。在本发明的这个方面,用于插入第二gsh中的可诱导盒优选地是如先前讨论的一种或多种主调节物。这些可诱导盒可以实现将细胞编程为特定谱系,并且将使用不同的可诱导盒,旨在指导分化为成熟细胞类型。构思了任何类型的成熟细胞,包括但不限于神经细胞、肌细胞、骨细胞、软骨细胞、上皮细胞、分泌细胞和/或血细胞。
本申请的发明人已经开发了一种实际上产生任何成熟细胞类型的快速、高效和可扩展方法。这种简单和便宜的方法将对再生医学具有特殊价值。先前的正向编程技术利用tet-on系统,但是企图将全部材料纳入一个载体/位点中(一体化tet-on)或尝试将可诱导盒插入一个aavs1等位基因并将控制系统插入另一个aavs1等位基因(dekelver等人,2010,genomeres.,20,1133-43和qian等人,2014,stemcells,32,1230-8)。令人惊讶地,此处开发和描述的双重gsh打靶方法具有许多不可预见的优点。在插入第一gsh中的基因和插入第二gsh中的可诱导盒的遗传序列之间不存在潜在的启动子干扰。第二,它允许从载体插入较大载货,因为需要在每个位点插入更少的材料。第三,该方法使安全插入的拷贝的数目最大化。其四,它能够实现更大的设计灵活性。最后,它允许插入额外遗传物质,包括报道基因和mirna开关。已经展示本发明的方法是从多能细胞生产成熟细胞的稳健和高效方式。
一旦基因已经插入第一gsh中并且包含转基因的可诱导盒已经插入第二gsh中,则可以培养多能干细胞以实现正向编程发生。对于正在使用的多能干细胞的类型,这些培养条件可以是特定的,或这些培养条件可以取决于最终的成熟细胞类型。无论使用何种培养条件,外源物质将控制可诱导盒内部遗传序列的表达;并且可以连续地供应并且随后撤回,旨在诱导转录,或当需要转录时供应,这取决于其作用模式,如先前讨论。
如果目的是对干细胞编程,可以有利的是,为细胞提供帮助分化的胞外激励,连同供应编码主调节物的可诱导盒。可以通过将主调节物或转录因子过量表达与胞外信号传导提示组合,增强细胞再编程策略。因此,可以通过调节涉及特定成熟细胞类型发育的主要信号传导级联,对促分化因子进行系统性筛选。这种例子见于实施例3中。
在一个方面,本发明提供一种从多能干细胞产生肌细胞的方法,所述方法包括步骤:
a)将编码转录调节蛋白的基因靶向插入第一遗传安全港位点中;和
b)将与诱导型启动子有效连接的myod1基因靶向插入第二遗传安全港位点中,其中所述诱导型启动子受转录调节蛋白调节;其中所述第一和第二遗传安全港位点是不同的,
并且在视黄酸存在下培养所述细胞。
myod1基因是编码生肌分化1蛋白的基因。优选地,视黄酸(ra)是全反式ra。
在另一个方面,本发明提供一种从表达myod1的多能干细胞产生肌细胞的方法,所述方法包括在视黄酸存在下培养所述细胞。
优选地,ra是全反式ra。优选地,细胞过量表达myod1。
在又一个方面,本发明提供一种从多能干细胞产生少突胶质细胞、肌细胞的方法,所述方法包括步骤:
a)将编码转录调节蛋白的基因靶向插入第一遗传安全港位点中;和
b)将与诱导型启动子有效连接的sox10基因靶向插入第二遗传安全港位点中,其中所述诱导型启动子受转录调节蛋白调节;其中所述第一和第二遗传安全港位点是不同的,
并且在视黄酸存在下培养所述细胞。
用于这种方法的细胞可以是动物细胞或人细胞。如果细胞属于动物,优选的是动物是如先前定义的家畜动物。
sox-10基因编码转录因子sox-10。优选地,视黄酸(ra)是全反式ra。
在本发明方法中所用细胞具有多能的情况下,通过表达主调节物,所得到的细胞可以是具有所需特性的谱系限制的特异性干细胞、祖细胞或成熟细胞类型。这些谱系特异性干细胞、祖细胞或成熟细胞可以按照任何合适方式使用。例如,如果对于细胞类型而言适宜,成熟细胞可以直接用于植入人体或动物身体。备选地,细胞可以形成研究用试验材料,包括药物对基因表达的影响和药物与特定基因相互作用。用于研究的细胞可以涉及使用具有功能未知的遗传序列的可诱导盒,旨在研究遗传序列的可控表达。另外,可以使得利用细胞产生大量所需物质如生长因子或细胞因子成为可能。
在不同方面,细胞可以用于组织工程中。组织工程需要生成组织,其可以用来替代人或动物的组织或甚至整个器官。组织工程方法是本领域技术人员已知的,但是包括使用应用细胞来产生组织/器官时的支架(胞外基质)。这些方法可以用来生成“人工”气管、膀胱、肝脏、胰、胃、肠、血管、心脏组织、骨、骨髓、粘膜组织、神经、肌肉、皮肤、肾或任何其他组织或器官。生成组织的方法可以包括增材制造(additivemanufacturing),另外称作三维(3d)打印,这可以包括直接打印细胞以产生组织。本发明因此提供一种使用如本发明任何方面中所述那样产生的细胞,来生成组织的方法。
使用根据本发明方法产生的细胞所生成的组织可以用于植入人体或动物身体。备选地,如果细胞来自动物,则组织可以用于体外/培养肉。用于培养肉的原代细胞类型是肌细胞。然而,这种组织可以涉及使用根据本发明方法产生的细胞类型的组合。这些类型可以是肌细胞(肌肉细胞)、血管细胞、血细胞和脂肪细胞(fatcell)。如果组织工程化的目的是用于培养肉,则细胞可以取自家畜动物。
出于多种原因,包括研究、基因治疗(包括基因疫苗)、产生体外疾病模型和生产非人类体内模型,也可以对不是多能干细胞的细胞实施本发明的方法。
本发明方法中所用的细胞可以因此是任何类型的成年干细胞;这些细胞是可以发育成许多类型、但非全部类型细胞的非特化细胞。成年干细胞是遍及身体存在的分裂以补充濒死细胞并且再生受损组织的未分化细胞。也称作成体干细胞(somaticstemcell),它们无多能性。已经在许多器官和组织中鉴定到成年干细胞,所述器官和组织包括脑、骨髓、外周血、血管、骨骼肌、皮肤、齿、心脏、肠、肝脏、卵巢上皮和睾丸。为了将细胞标记为成体干细胞,技术人员必须展示,单个成年干细胞可以生成一系列遗传相同的细胞,所述细胞随后产生组织的全部适宜的分化细胞类型。为了实验上确认推定性成年干细胞的确是干细胞,细胞必须在培养中产生这些遗传相同的细胞,或这些细胞的纯化群体必须在植入动物后重新构成组织。合适的细胞类型包括但不限于神经、间充质和内胚层干细胞和前体细胞。
备选地,使用的细胞可以是成熟细胞类型。这类细胞分化和特化并且不能够发育成不同的细胞类型。成熟细胞类型包括但不限于神经细胞、肌细胞、骨细胞、软骨细胞、上皮细胞、分泌细胞和/或血细胞。成熟细胞类型可以是来自人体或动物身体的任何细胞。
成体干细胞和成熟细胞类型可以根据本发明修饰并且随后用于多种应用如基因治疗或遗传接种。基因治疗可以定义为将外来dna人为插入治疗目的细胞的胞核。这种定义包括向细胞供给一个基因或多个基因以提供野生型形式的错误基因、添加干扰靶基因表达(其可能有缺陷)的rna分子的基因、供给自杀基因(如将无害前药更昔洛韦(gcv)转化成细胞毒药物的酶—单纯疱疹病毒胸苷激酶(hsv-tk)和胞嘧啶脱氨酶(cd))、用于免疫疗法或癌疗法(包括细胞过继免疫疗法)的dna疫苗和用于治疗目的的任何其他向细胞供给基因。
一般而言,本发明的方法可以用于插入目的遗传序列以便在细胞中转录,优选地表达,特别地在dna疫苗中。dna疫苗一般编码传染性生物的dna的修饰形式。将dna疫苗施用至受试者,其中它们随后表达传染性生物的选定蛋白质,从而启动一般是保护性的针对该蛋白质的免疫应答。dna疫苗还可以编码癌症免疫治疗方案中的肿瘤抗原。
dna疫苗可以包含编码治疗或预防众多病症的抗原的核酸序列,其中所述病症包括但不限于癌症、变态反应、中毒和病原体感染,所述病原体例如是但不限于真菌;病毒,包括人乳头瘤病毒(hpv)、hiv、hsv2/hsv1、流感病毒(a、b和c型)、脊髓灰质炎病毒、rsv病毒、鼻病毒、轮状病毒、甲型肝炎病毒、麻疹病毒、副流感病毒、腮腺炎病毒、水痘带状疱疹病毒、巨细胞病毒、eb病毒、腺病毒、风疹病毒、人t细胞淋巴瘤i型病毒(htlv-i)、乙型肝炎病毒(hbv)、丙型肝炎病毒(hcv)、丁型肝炎病毒、痘病毒、zika病毒、马堡病毒和埃博拉病毒;细菌,包括脑膜炎球菌(meningococcus)、流感嗜血杆菌(haemophilusinfluenza)(b型);和寄生性病原体。dna疫苗可以包含编码来自任何合适病原体的抗原的核酸序列。抗原可以来自造成人疾病或兽医疾病的病原体并且尤其可以来自病毒性病原体。
插入gsh中的dna疫苗还可以包含编码肿瘤抗原的核酸序列。肿瘤相关抗原的例子包括但不限于癌抗原,如mage家族成员(mage1、2、3等)、ny-eso-i和ssx-2、分化抗原如酪氨酸酶、gp100、psa、her-2和cea、突变的自我抗原和病毒性肿瘤抗原如来自致瘤hpv类型的e6和/或e7。具体肿瘤抗原的其他例子包括mart-i、melan-a、p97、β-hcg、gainac、mage-i、mage-2、mage-4、mage-12、mucl、muc2、muc3、muc4、muc18、cea、ddc、pla、epcam、黑素瘤抗原gp75、hker8、高分子量黑素瘤抗原、kl9、tyrl、tyr2、pmel17基因家族成员、c-met、psm(前列腺黏蛋白抗原)、psma(前列腺特异性膜抗原)、前列腺分泌蛋白、甲胎蛋白、ca125、ca19.9、tag-72、brca-1和brca-2抗原。
插入的遗传序列可以产生其他类型的治疗用dna分子。例如,在受试者患有由功能基因的功能障碍形式所致的遗传病情况下,这类dna分子可以用来表达该基因。这类疾病的例子包括杜兴氏肌营养不良、囊性纤维化、gaucher病和腺苷脱氨酶(ada)缺乏症。其中基因治疗可能有用的其他疾病包括炎性疾病、自身免疫性、慢性和传染性疾病,包括这类病症,如aids、癌症、神经系统疾病、心血管疾病、高胆固醇血症、多种血液病症,包括多种贫血、地中海贫血和血友病,和肺气肿。为了治疗实体瘤,可以表达编码有毒肽(即,化疗药如蓖麻毒蛋白,白喉毒素和眼镜蛇毒液因子)的基因、肿瘤阻抑基因如p53、编码对转化性癌基因反义的mrna序列、抗肿瘤肽如肿瘤坏死因子(tnf)和其他细胞因子或转化性癌基因的反式显性负突变体的基因。
还构思了其他类型的治疗用dna分子。例如,可以插入转录成有活性、非编码性rna形式(例如小干扰性rna(sirna))的dna分子。本发明的方法因此扩展至使用可诱导盒中的非编码性rna,敲低内源性基因表达或敲除内源基因的方法。
因此,本发明的方法可以用来特异性和稳定插入遗传序列于可诱导盒中,其可以可控转录。这在成体干细胞和成熟细胞类型方面具有众多优点。它允许更紧密调节的基因治疗方案,从而确保不破坏关键基因并且若出现任何不良作用,则允许关闭可诱导盒的表达。它还允许敲低或敲除紧密调节的内源性基因,以探查基因功能和发育。
本发明扩展至通过本发明方法产生的细胞。这些细胞可以定义为在第一基因组安全港位点被修饰,以包含转录调节蛋白,和在第二遗传安全港位点被修饰,以包含与受转录调节蛋白调节的诱导型启动子有效连接的遗传序列。两个gsh是不同的和有区别的。优选地,细胞在两个插入位点纯合。全部元件如前所述。
根据本发明的任何方法产生的细胞已经应用于诊断方法和治疗方法中。细胞可以体外用来研究细胞发育、为新药物提供测试系统、能够筛查待开发的方法、审查治疗方案、提供诊断性检验等。这些用途形成本发明的部分。备选地,出于诊断或治疗目的,细胞可以植入人类或动物患者。本发明中还包括细胞在疗法中的用途。细胞可以是同种异基因的(即取出成熟细胞、修饰并返回相同个体)或来自供体(包括干细胞系)。
本文中提及的全部文献因而通过引用的方式并入。
序列
aavs1-ncbigenbanks51329.1
seqidno1:tet0219n序列
seqidno2:hrosa插入位点基因组序列
seqidno3:stdtetr-nls(核苷酸)和seqidno4-stdtetr-nls(氨基酸)
seqidno5:opttetr-nls(核苷酸)和seqidno6-opttetr-nls(氨基酸)
seqidno7至80:来自表3的引物。
seqidno81:图18baavs1fwd;seqidno82:图18baavs1rev
seqidno83:图18b示踪剂fwd;seqidno84:图18b示踪剂rev
seqidno85:图19ehipol3fwd;seqidno82:图19ehipol3rev
这是hrosa26插入位点的基因组序列;它包括5’同源性臂、切割位点(粗体)和3’同源性臂:(seqidno2)
gctcgaaaccggacggagccattgctctcgcagagggaggagcgcttccggctagcctcttgtcgccgattggccgtttctcctcccgccgtgtgtgaaaacacaaatggcgtattctggttggagtaaagctcctgtcagttacgccgtcgggagtacgcagccgcttagcgactctcgcgttgccccctgggtggggcgggtaggtaggtggggtgtagagatgctgggtgtgcgggcgcggccggcctcctgcggcgggaggggagggtcagtgaaatcggctctggcgcgggcgtcctcccaccctccccttccttcgggggagtcggtttacccgccgcctgcttgtcttcgacacctgattggctgtcgaagctgtgggaccgggcccttgctactggctcgagtctcacatgagcgaaaccactgcgcggggcgcgggggtggcggggaggcgggcgttggtacggtcctccccgaggccgagcgccgcagtgtctggccccgcgcccctgcgcaacgtggcaggaagcgcgcgctggaggcgggggcgggctgccggccgagacttctggatggcggcggccgcggctccgccccgggttcccaccgcctgaagggcgagacaagcccgacctgctacaggcactcgtgggggtgggggaggagcgggggtcggtccggctggtttgtgggtgggaggcgcttgttctccaaaaaccggcgcgagctgcaatcctgagggagctgcggtggaggaggtggagagaaggccgcacccttctgggcagggggaggggagtgccgcaatacctttatgggagttctctgctgcctcccgtcttgtaaggaccgccctgggcctggaagaagccctccctcctttcctcctcgcgtgatctcgtcatcgcctccatgtcgagtcgcttctcgattatgggcgggattcttttgcctaggcttaaggggctaacttggtccctgggcgttgccctgcaggggagtgagcagctgtaagatttgaggggcgactccgattagtttatcttcccacggactagagttggtgtcgaggttattgtaataagggtggggtagggaaatggagcttagtcattcacctggggctgattttatgcaacgagactgcggattatcactacttatcatttttggagcatttttctagagacagacataaagcatgatcacctgagttttataccatttgagacccttgctgcaccaccaaagtgtagcatcaggttaaatcttaatagaaaaattttagcttttgcttgagaaaccagtgcttccctccctcaccctctctccccaggctctctacccctttgcatccctaccaggcatcttagcaactctcactcatacttgatcccattttccatttgttgtacttgctcctctagtattcagacatagcactagctttctccctctcttgatcttgggtagcctggtgtctcgcgaaaccagacagattggttccaccacaaattaaggcttgagctggggcttgactcttacccagcagtgcttttattcctccctagttcacgttcttaaatgtttatcttgattttcattttatcctttttccttagctgggattctgtccctgaccgtcttcacagtccaggtgatcttgactactgctttacagagaattggatctgaggttaggcaacatctccctttttcttcctctaaatacctctcatttctgttcttaccagttagtaactgatctcagatgcctgtgtgatagcttcc
stdtetr-nls:(seqidno3和4)
含有n末端sv40核定位信号(nls,以灰色突出显示)的四环素敏感阻抑蛋白(tetr)的核苷酸序列和氨基酸序列。在密码子优化之前或之后报道序列(分别是stdtetr和opttetr)。点表示在opttetr中引入的同义突变。
优化的tetr:opttetr-nls的序列(seqidno5和6)
现在将相对于以下非限制性实施例描述本发明:
实施例
实施例中所用的材料和方法:
hpsc维持培养和胚层分化
进行无饲养细胞和无血清hesc(h9系;wicell)和hipsc(cheung等人,nat.biotechnol.30,165–173(2012))培养。简而言之,将细胞铺种在明胶/mef培养基包被的培养皿上[mef培养基由高级dmem/f12(90%,gibco)、胎牛血清(10%,gibco)、l-谷氨酰胺(1mm,gibco)、2-巯基乙醇(0.1mm,sigma-aldrich)和青霉素/链霉素(1%,gibco)组成]并在补充有10ng/ml活化素-a和12ng/mlfgf2的化学成分确知培养基[cdm,由imdm(50%,gibco)、f12(50%,gibco)、浓缩脂质(100x,gibco)、单硫代甘油(450μm,sigma-aldrich)、胰岛素(7μg/ml,roche)、转铁蛋白(15μg/ml,roche)、牛血清白蛋白级分v(5mg/ml)和青霉素/链霉素(1%)组成]中培养。每5-6天,使用胶原蛋白酶,将细胞在小团块中传代。
在贴壁hesc培养物中根据先前发表的内胚层、侧板中胚层和神经外胚层定向分化方案,诱导hpsc分化成胚层(touboul,t.等人hepatology51,1754–1765(2010);cheung等人,(2012)和douvaras,p.等人stemcellreports3,250–259(2014))。简而言之,通过将hpsc在补充有fgf2(20ng/ml)、活化素-a(100ng/ml)、bmp4(10ng/ml,markomarkohyvonen,生物化学系,剑桥大学)和ly-294002(10μm,promega)3的cdm-pva(无胰岛素)中培养3天,衍生定形内胚层。为了衍生神经外胚层,将hpsc在补充有sb-431542(10μm,tocris),ldn-193189(0.1μm,tocris)和ra(0.1μm,sigma)4的cdm-bsa中培养6天。通过将hpsc在补充有fgf2(20ng/ml)、10ng/mlbmp4(r&d)和ly294002(10μm)的cdmpva中培养36小时并且在补充有fgf2(20ng/ml)和bmp4(50ng/ml)的cdm-pva中后续培养3.5天,获得侧板中胚层。
hesc的分化。传代后贴壁培养hesc48小时,引发分化。通常每日进行培养基更换,并且针对细胞密度调整体积。使用本领域先前描述的方法,获得成熟细胞类型。获得的成熟细胞类型包括神经细胞、骨细胞、软骨细胞、平滑肌、心脏成纤维细胞、心肌细胞、肠、胰、肝细胞、胆管细胞或肺。
基因打靶构建体和分子克隆
此处描述hrosa26grna和cas9n表达质粒的设计和构建:基于crispr/cas9n的特异性靶向hrosa26基因座并使用同源重组插入可诱导盒的策略。为了在正确的整合位点诱导基因组dsb,设计了crispr/cas9切口酶系统。与被单一grna导引至其基因组靶位点的常用野生型cas9核酸酶相反,d10a突变体cas9切口酶(cas9n)受一对适当设计的在靶dna的两条链上同时引入单链切口的grna指导。这个策略有效地加倍基因组编辑所需要的碱基的数目并且因而增加特异性。基于网络的软件“crispr设计工具”用来定义接近于整合位点的crrna指导型核酸酶的潜在靶位点。在靶位点周围的250bp序列段之内(实际整合位点的每个位点上125bp),最高命中产生一对总体上达到“高质量”评分97、预测无脱靶效应的grna。从头合成grna[grna-a5’-gtcgagtcgcttctcgatta-(tgg)-3’和grna-b5’-ggcgatgacgagatcacgcg-(agg)-3’(括号中的pam位点)并且连接入表达载体中。最终的质粒分别编码两种grna的任一者和cas9nd10a-突变体(图20和图21)。
构建供体质粒,其充当模板dna以促进cas9n所致dsb的同源性指导修复。通过高保真pcr扩增产生两条hrosa26同源性臂。从h9hesc分离的基因组dna充当模板。5’和3’同源性臂在长度上分别是904bp和869bp。随后将二者插入puc19载体的多克隆位点中。为了打靶hrosa26基因座,将细胞用该质粒、两种grna/cas9n构建体和egfp供体质粒转染(图22)。
通过以下方式构建pr26_cag-rtta打靶载体(图23):将第三代rtta的编码性序列(pcr扩增自plvx-tet3g)克隆入pr26_cag-egfp的bamhi/mlui位点,因此替换egfp序列。aavs1zfn表达质粒是kosukeyusa博士(wellcome-trustsangerinstitute)的慷慨馈赠。由gibsonassembly(newenglandbiolabs)构建诱导型egfpaavs1打靶载体,其中将三个插入物连接入puc19载体(thermofisherscientific)多克隆位点的ecori/hindiii位点:第一插入物包含上游aavs1同源性臂、剪接受体、t2a位点和嘌呤霉素耐药性盒(pcr扩增自ptre-egfp;addgene22074,由rudolfjaenisch保藏)。第二插入物含有诱导型tre3g启动子(pcr扩增自plvx-tre3g)。第三插入物包含egfp表达盒和aavs1下游同源性臂(pcr扩增自ptre-egfp;addgene22074,由rudolfjaenisch保藏)。所产生的质粒称作paav_tre-egfp(图32)。通过以下方式构建paav_tre-ngn2和paav_tre-myod1(图33)打靶载体:将ngn2和myod1编码性序列(ngn2:pcr扩增自plvx-tre-ngn2,获赠自oliverbrüstle;myod1:pcr扩增自市售cdna质粒,openbiosystemsmhs6278-202832821,登录号:bc064493,克隆id:5022419)分别克隆入paav_tre-egfp的spei/ecori位点,因此替换egfp序列。
使用相似方法还产生其他质粒,并且所用的全部质粒均在图20至图33中描述。这些质粒系创建或慷慨捐赠。实施例中使用的质粒包括(按图20–图33的顺序):pspcas9n(bb)_r26-r、pspcas9n(bb)(预测这两种质粒的组合在染色体3上thumpds3-as1的外显子1和2之间的内含子中(rosa26基因座)诱导特定双链断裂)、_r26-lpr26_cag_egfp,pr26_cag_rtta、pzfn-aavs1-l-eld(锌指核酸酶(左))、pzfn-aavs1-r-kkr(锌指核酸酶(右))、paav_cag_egfp(供体)、pr26-neo_cag-opttetr(密码子优化的tetr的hrosa26打靶)、paav-puro_ikd(诱导型shrna的aavs1打靶)、paav-neo_cag-cas9(cas9的aavs1打靶)、paav-puro_siko(诱导型grna的aavs1打靶)、paav-puro_siko-2to(诱导型grna的aavs1打靶,启动子中具有2个tet操纵子的形式)、paav_tre-egfp(egfp诱导型过量表达,附加的)和paav_tre-myod1(肌肉的myod1诱导型过量表达)。
基因打靶
通过核转染法进行hrosa26基因座和aavs1打靶,以便基因敲低和敲除。将人多能干细胞(psc)用trypleselect(gibco)解离成单个细胞,并且使用lonzap3原代细胞4d-nucleofectorx试剂盒和lonza4d-nucleofector系统的循环ca-137,核转染2x106个细胞(100μl反应体积;总计12μgdna,其在两种grna/cas9n质粒和打靶载体之间等分)。将核转染的hpsc铺种在照射过的多药耐药性(dr4)小鼠胚胎成纤维细胞上并且在补充有fgf2(4ng/ml,生物化学系,剑桥大学)的ksr培养基[由高级dmem/f12(80%)、敲除血清替换物(20%,gibco)、l-谷氨酰胺(1mm)、2-巯基乙醇(0.1mm)和青霉素/链霉素(1%)组成]中培养。在核转染之前和之后添加y-27632(5μm,tocris)持续24小时以促进细胞存活。在3-6天后,通过添加g418(50μg/ml,sigma-aldrich)持续7-10天,选择耐新霉素的hpsc。随后,将独立的克隆挑出,在无饲养细胞条件下扩充并通过基因分型法最终分析。
还通过脂转染进行aavs1基因座打靶。将人psc在无饲养细胞条件下接种于6孔板中,并且在传代后48小时转染。转染在补充有脂转染胺2000(10μl/孔,thermofisherscientific)和总计4μgdna(在两种aavs1zfn质粒和打靶载体之间等分等分)的opti-mem(gibco)中进行24小时。在3-5天后,通过添加嘌呤霉素(1μg/ml,sigma-aldrich)持续5-8天,选择抗性hpsc。随后,将独立的克隆挑出,扩充并通过基因分型法分析。抗生素耐药性可以用来选择克隆系。
通过基因组pcr筛选来自打靶实验的耐药hpsc克隆,以核实位点特异性可诱导盒整合、确定靶向的等位基因的数目并排除脱靶整合。用longamptaqdna聚合酶(newenglandbiolabs)进行pcr。表2报道了用于多种打靶载体的引物组合。表1中总结了全部打靶实验的结果。通过标准g显带技术(medicalgeneticsservice,剑桥大学医院)进行染色体组型分析。为了制备靶向的人psc供染色体分析,将细胞在补充有y-27632(5μm,tocris)和karyomaxcolcemid(100ng/ml,gibco)的新鲜培养基中在+37℃温育4小时。随后,将细胞作为单个细胞收获、洗涤并沉淀。通过用低渗0.055mkcl溶液处理5-10分钟,实现胞核肿胀和染色体展开。最后,将细胞用甲醇和冰醋酸(比率3:1)固定。
对于optikd,通过如先前所描述的脂转染法进行aavs1打靶。简而言之,将hpsc无饲养细胞接种在6孔板中并且在细胞传代后48小时,使用opti-mem培养基(gibco)中的10μl/孔脂转染胺2000,以4μgdna(在两种aavs1zfn质粒和打靶载体之间等分)转染24小时,全部操作根据生产商的说明书进行。在4天后,将1μgml-1嘌呤霉素添加至培养基,并且在7-10天选择后,挑出独立的克隆并扩充。
对于单个位点optiko,通过核转染法进行aavs1打靶。使用accutase(gibco),将采用10μmy-27632(tocris)预处理16小时的hesc解离成2-8个细胞的团块,并且使用lonzap3原代细胞4d-nucleofectorx试剂盒和lonza4d-nucleofector系统上的循环ca-137,用总计12μgdna(两种zfn质粒各自4μg,并且两种靶载体各自2μg)以100μl核转染2x106个细胞,全部操作均根据生产商的说明进行。将核转染的hesc铺种在照射过的dr4(嘌呤霉素和新霉素耐药性)小鼠胚胎成纤维细胞的滋养层上并且在补充有4ngml-1fgf2和10μmy-27632的ksr培养基中培养(这持续仅前24小时)。在4天后,将携带嘌呤霉素耐药基因和新霉素耐药基因的hpsc集落用25μgml-1遗传霉素(硫酸g418,gibco)和0.5μgml-1嘌呤霉素选择7-10天。将独立的克隆随后挑出并且在无饲养细胞条件下扩充。
用aavs1zfn或rosa26crispr/cas9n对通过脂转染(aavs1基因座)或核转染法(rosa26基因座)打靶载体(如上文所述),产生aavs1-egfp、rosa26-egfp、rosa26-stdtetr、rosa26-opttetr和rosa26-egfpd2hesc。2μgml-1杀稻瘟素s-hcl(gibco)用于pr26-bsd_cag-egfpd2质粒。他处描述携带rosa26-rtta和aavs1-tre-egfp转基因的诱导型egfp过量表达hesc的产生。简而言之,首先通过用rosa26crispr/cas9n质粒对pr26-neo_cag-rtta进行核转染,随后通过用aavs1zfn质粒对paavpuro_tre-egfp进行脂转染,对细胞依次基因打靶。
通过基因组pcr筛选基因打靶的hpsc克隆系,以核实位点特异性打靶、测定已靶向的等位基因的数目并且排除打靶质粒的脱靶整合(参见图16a)。
可诱导盒过量表达
通过添加盐酸多西环素(sigma-aldrich)至培养基,诱导过量表达可诱导盒(分别是egfp、ngn2、myod1和olig2-sox10)。除非另外说明,否则按终浓度1μg/ml使用多西环素。含有多西环素的培养基保持避光,并且每24小时更换。在本文中,表达egfp的细胞称作opti-egfp,表达ngn2的那些细胞称作opti-ngn2,表达myod1的细胞称作opti-myod1并且表达olig2-sox10的细胞称作opti-olig2-sox10。
诱导型基因敲除和敲低
除非图注或实施例中另外描述,否则按1μgml-1使用盐酸四环素(sigma-aldrich)以诱导基因敲低或敲除。
神经元的诱导
将多能opti-ngn2细胞用tryple解离成单个细胞并按12孔板每孔75,000个细胞的密度铺种在matrigel(35μg/cm2,scientificlaboratorysupplies)涂覆的培养皿上。在分瓶后24-48小时启动正向编程。除非另外说明,否则在补充有glutamax(100x,gibco)、非必需氨基酸(100x,gibco)、2-巯基乙醇(50μm)、青霉素/链霉素(1%)和多西环素(1μg/ml)的dmem/f12(gibco)中进行诱导。在诱导2天后,将培养基转换成补充有glutamax(100x)、b27(50x,gibco)、bdnf(10ng/ml,peprotech)、nt3(10ng/ml,r&dsystems)、青霉素/链霉素(1%)和多西环素(1μg/ml)的神经基础培养基。
骨骼肌细胞的诱导
将多能opti-myod1细胞用tryple解离成单个细胞并按12孔板每孔100,000个细胞的密度铺种在明胶/mef培养基涂覆的培养皿上。在分瓶后24-48小时启动正向编程。除非另外说明,否则在补充有l-谷氨酰胺(2mm)、2-巯基乙醇(50μm)、青霉素/链霉素(1%)、胰岛素(7μg/ml)、全反式视黄酸(1μm,sigma-aldrich)和多西环素(1μg/ml)的dmem(sigma-aldrich)中进行诱导。在诱导5天后,向培养基补充chir99021(3μm,tocris)和热灭活的马血清(2%,gibco)以增强成熟。
少突胶质细胞的诱导
在明胶/mef涂覆的培养皿上培育多能olig2-2a-sox10opti-oxhpsc成集落。在诱导开始之前,将它们用sb和ldn过夜处理。次日,在补充有多西环素(1μg/ml)和ra(0.1μm)的cdm中启动诱导。诱导后一天,将细胞在补充有ra(0.1μm)、pm(1μm)和y-27632(5μm)、pdgfaa(20ng/ml,peprotech)、fgf2(5ng/ml)的cdm中分到pdl/层粘连蛋白涂覆的培养皿(12孔板每孔100,000个细胞)上。次日,将细胞切换成少突胶质细胞培养基,所述培养基由dmem/f12组成,补充有glutamax(100x)、非必需氨基酸(100x)、2-巯基乙醇(1000x)、青霉素-链霉素(100x)、n2补充物(100x)、b27补充物(50x)、胰岛素7μg/ml(markohyvonnen)、t360ng/ml(sigma)、生物素100ng/ml(sigma)、db-camp1μm(sigma)。向少突胶质细胞培养基补充dox(1μg/ml)、pdgfaa(20ng/ml)、fgf2(5ng/ml)、ra(0.1μm)和pm(1μm)。诱导后七天,撤回ra和pm。为了保持诱导细胞处于增殖状态,在促分裂原pdgfaa和fgf2持续存在下,将细胞每4天传代(24孔板每孔75,000个细胞)。为了分化增殖性少突胶质细胞前体,撤回pdgfaa和fgf2。添加人重组nt3(5ng/μl,r&dsystems)以增强细胞存活。
定量实时pcr(qpcr)
使用genelute哺乳动物总rna微量制备试剂盒和柱上dna酶i消化套装(sigma-aldrich)提取rna。用maxima第一链cdna合成试剂盒(thermofisherscientific)进行cdna合成。appliedbiosystemssybrgreenpcr主混合物用于qpcr。在appliedbiosystems7500快速pcr仪上运行样品。全部样品以技术性重复进行分析并且对持家基因胆色素原脱氨酶1(pbgd)归一化。用δδct方法分析结果。引物序列参见表3。
流式细胞术
为了分析egfp表达,用trypleselect(gibco)在37℃持续5-10分钟,收获细胞,以获得单细胞悬液。用pbs洗涤后,将细胞重悬于补充有dapi(10μg/ml)的冰冷pbs中并且在冰上温育5分钟。使用cyanadp流式细胞仪分析细胞,以确定活细胞(dapi阴性)的egfp表达水平。为了染色和分析肌球蛋白重链表达,将细胞用trypleselect(如egfp表达分析那样)收获,用pbs洗涤一次并用cytofix/cytoperm液(bdbiosciences)固定和透化。随后,将细胞在补充有3%牛血清白蛋白(bsa)的perm/洗涤缓冲液(bdbiosciences)中洗涤并在4℃封闭过夜。在perm/洗涤缓冲液中在4℃于黑暗下实施pe缀合的抗-myh抗体(表4)染色1小时。用perm/洗涤缓冲液洗涤三次后,用cyanadp流式细胞仪分析细胞,以确定mhc表达水平。用flowjo(v10)和graphpadprism(v6)进行数据分析。
蛋白质印迹法
用补充了完全蛋白酶抑制剂(roche)的cellyticm(sigma-aldrich)提取全细胞蛋白质,并且随后通过使用蛋白质快速定量试剂盒(sigma-aldrich)定量。用nupagelds样品缓冲液和4-12%nupagebis-tris预制凝胶(invitrogen)进行蛋白质电泳。在pvdf上转移蛋白质后,将膜用补充有0.05%tween-20(pbst)4%乳的pbs在室温封闭1小时,并且在pbst4%乳中与第一抗体温育过夜。将膜用pbs洗涤,在pbst4%乳中与hrp缀合的第二抗体(sigma-aldrich)温育,与pierceecl2蛋白质印迹底物(thermofisherscientific)温育并且对x光superrx胶片曝光(fujifilm)。
免疫细胞化学
将细胞在4%多聚甲醛中(稀释于pbs中)室温固定20分钟并切随后用pbs洗涤三次。将细胞随后用10%驴血清(sigma-aldrich)封闭并且在室温用0.3%tritonx-100(稀释于pbs中)透化20分钟。随后,将细胞在2%驴血清和0.1%tritonx-100中(稀释于pbs中)与适当稀释的第一抗体(补充性实验步骤)于4℃温育过夜。在着染表面抗原pdgfra、a2b5和o4时,全部步骤自始至终省略triton-x。用pbs洗涤三次后,将细胞在补充有1%驴血清的pbs中于室温与相应的荧光团缀合的驴第二抗体(alexafluor488、555、568和/或647)温育1小时。胞核用4',6-二脒基-2-苯基吲哚(dapi,thermofisherscientific)可视化。使用zeisslsm700共聚焦显微镜(leica),对egfp表达和免疫染色成像。通过使用倒置olympusix71荧光显微镜,在3个生物学重复的3个视野的至少50个随机选择的dapi阳性细胞中确定βiii-微管蛋白表达,计算βiii-微管蛋白阳性细胞百分数。
用graphpadprism(v6)进行统计分析。图注中描述重复的数目、使用的统计检验和试验结果。除非另外说明,否则数据表示为均数±sem。
实施例1:egfp双重打靶
为了在hpsc中开发诱导型过量表达平台,我们将tet-on系统的两个组件依次打靶入两个不同的gsh。通过使用基于crispr/cas9n的打靶策略,将组成型表达的第三代rtta打靶至人rosa26(hrosa26)基因座中,并且将诱导型egfp可诱导盒插入aavs1中(图1a;图4a–c)。hrosa26和aavs1打靶均高效(图4d-f,表1)并且不影响hpsc基因组稳定性、自我更新和分化(数据未显示),因此驳斥rtta依赖的细胞毒性。
我们随后选择双重gsh打靶的克隆,所述克隆携带两种可诱导盒各自的一个或两个拷贝(图5a)。与杂合rtta表达相比时,诱导后,rtta纯合打靶产生大约两倍较高的rtta蛋白水平(图5b),并且还产生显著增加的egfp水平(图5c-5e)。另外,与具有杂合打靶的系相比,具有诱导型egfp盒纯合打靶的克隆显示更高和更均匀的egfp水平(图5c-图5e)。重要地,全部正确靶向的系均显示稳健的诱导型egfp表达,与强力的组成型cag启动子相比,所述表达至少二十倍更高(图1b、图5c-e)。总体上,这些结果支持我们起初的假设:靶向两个tet-on系统元件的两个拷贝将导致诱导后表达最大。大约诱导后四天,达到egfp峰水平,并且当撤回多西环素时,表达迅速逆转(图1c)。另外,可以通过调节多西环素剂量滴定egfp表达(图1d)。重要地,诱导型egfp表达不仅在hpsc中十分高效,在分化成胚层期间也十分高效(未显示彩色摄影数据,数据在图6a-6d上)。最后并且与已知的第三代tet-on系统紧密转录性控制一致,在多西环素不存在的情况下,不存在egfpmrna或蛋白质的可检测背景表达,分别如流式细胞术和qpcr所测定(图1b,图6d)。总之,这些结果确立了,tet-on系统的双重gsh打靶是在hpsc及其衍生物中最佳表达可诱导盒的有力策略。
实施例2:从hesc和hipsc衍生兴奋性皮质神经元
先前研究已经显示,可以在hpsc中通过慢病毒过量表达任何促神经元bhlh因子(ascl1、ngn2或neurod1),轻易地衍生这些细胞。因此,我们生成了opti-ngn2hpsc(图2a,表1)。ngn2诱导导致多能性因子快速下调(图7)及神经元转录程序启动(图2b)。在早至诱导后三天,诱导的细胞显示出神经元突起(数据未显示)。在一周后,全部细胞均展示神经元形态并且表达泛神经元标志蛋白,如βiii-微管蛋白和map2(图2c)。定量rt-pcr揭示强力诱导常见的前脑标志物(如brn2和foxg1)和包含gria4和vglut2的谷氨酸能神经元(图2b),从而提示兴奋性皮质神经元身份。总体上,这些结果显示,与传统hpsc分化方案相比,产生神经元的速度和效率大幅度改善,并且相对于转分化和基于慢病毒的正向编程方案,效率和纯度实质性地增加。用opti-ngn2hipsc获得相似结果,从而确认这种方法的稳健性。最后,在延长的opti-ngn2hpsc培养期间(>25代,图2c),我们未观察神经元诱导效率的任何降低。总之,我们的结果显示,opti-ngn2hpsc可以作为无穷无尽的来源用于不受限制、高度可扩展、快速、单步骤、无病毒和近乎确定地产生神经元。
实施例3:产生骨骼肌细胞
已知在多种体细胞类型中过量表达时,转录因子myod1诱导生肌转分化,然而,hpsc发生myod1诱导的生肌正向编程的能力目前存在争议。我们生成了opti-myod1hpsc(表1),但是我们指出,多西环素处理后诱导myod1表达导致细胞在广阔范围的培养条件下3-5天内几乎完全死亡,其中先前提出所述培养条件促进hpsc转化成骨骼肌细胞。由于广泛地确立了,可以通过转录因子过量表达与胞外信号传导提示组合增强细胞再编程策略,所以我们通过调节参与原条形成、体节发生和肌生成的主要信号传导级联,系统性筛选了促生肌因子。我们发现,截止诱导后第5天,添加全反式视黄酸(ra)联合myod1过量表达导致向肌细胞生成素和肌球蛋白重链(mhc)双阳性肌细胞的快速和近乎完全转化。ra的影响是浓度依赖的并且由ra-受体同工型rarα和rarβ介导,这与发育性肌生成期间的ra受体表达模式一致(图8)。认为这种影响与myod1过量表达的机制无关。诱导的骨骼肌细胞展示出常见的纺锤体样、细长形态,发生广泛的细胞融合并且在mrna水平和蛋白质水平均显示强烈的生肌标志物表达(图3b、图9a–9c)。添加纳摩尔浓度的乙酰胆碱(ach)或选择性ach受体激动剂卡巴胆碱导致完全的肌纤维收缩,从而显示诱导的肌细胞的功能。用opti-myod1hipsc获得相似的结果(数据未显示)。重要地,生肌诱导效率在延长的培养时间(>50代,图3d)内不下降,因此显示这种方法的稳健性和重现性。最后,我们指出,myod1诱导型盒的水平与转化效率正相关,这突出显示稳健递送基因的重要性和这种方法胜过慢病毒介导再编程方案的优效性(图10)。总之,opti-myod1正向编程策略比最新的hpsc分化成骨骼肌细胞方案快大约七倍并且效率高大约五倍。与先前的正向编程方案相比(tanaka,a.等人plosone8,e61540(2013)和abujarour,r.等人stemcellstransl.med.3,149–60(2014)),它更高效(>95%,相比于30-80%而言),不含随机插入的可诱导盒,化学成分确知、完全可重复并且可扩展性更大。
这些结果显示,这种在hpsc中控制可诱导盒表达的方法可以作为无穷无尽的来源用于高通量和大规模生产均匀的细胞群体。目的靶细胞的诱导速度和纯度目前是其他方法无可匹敌的。
实施例4:产生少突胶质细胞前体和少突胶质细胞:
携带单独或与双顺反子表达盒形式的olig2组合的诱导型sox10的opti-oxhpsc。尽管10天诱导后,仅用sox10诱导的细胞稳健表达少突胶质细胞前体(opc)标志物o4,但是这些细胞没有进一步分化成表达髓鞘质的细胞并且逐渐地死亡。相反,在诱导后20天,olig2-sox10双重过量表达的细胞轻易地从o4-阳性祖细胞阶段进展为成熟的cnp/mbp-阳性表型。另外,额外的标志蛋白质表达分析证实,在补充有促分裂原pdgfaa和fgf2的少突胶质细胞培养基(douvaras等人,2014)中诱导的opti-olig2-sox10hpsc首先通过opc样阶段,其中它们具有高度增殖性并且其中它们共表达pdgfra、a2b5和o4。这些细胞具有高度增殖性并且可以通过在促分裂原存在下培养它们,维持至少三代(图12b)。我们因此将这些细胞命名为i-opc,即诱导的opc。值得注意地,在撤回促分裂原后和多西环素持续存在时,i-opc在大约一周内轻易分化为表达能够形成髓鞘的主要髓鞘蛋白cnp、plp、mag、mog和mbp(图12c-12d)的成熟少突胶质细胞(数据未显示)。总体上,这些结果显示,本发明允许形成新的、稳健的和快速的生成少突胶质细胞前体和少突胶质细胞的hpsc正向编程方案。
表1:基因分型结果总结:
(a)不正确打靶:无打靶证据(在5’-和3’-整合pcr中缺少条带和在基因座pcr中存在wt条带)或有打靶证据,但是5’-或3’-整合pcr的大小不正确。
(b)具有质粒的附加随机整合的正确中靶整合(3’-骨架pcr中的条带)。
(c)正确中靶整合(het,杂合;hom,纯合)。
(d)具有正确中靶整合(无附加脱靶整合)的克隆的百分数
(e)具有正确中靶整合(有或无附加脱靶整合)的克隆的百分数
*三个数字来自三个在hesc中不同的打靶实验。
表2:
用于基因分型pcr的引物列表
表3:定量pcr引物的列表
表4:抗体列表
实施例5:tet-on可诱导敲低系统
在hpsc中开发优化的可诱导敲低平台。
我们生成了其中egfp转基因可以按照可诱导方式沉默的hesc系(图14b)。为此,我们(1)将cag-tetr表达盒靶入rosa26基因座;和(2)将cag-egfp转基因加诱导型egfpshrna盒靶入aavs1基因座(图14a,b)。表达更高水平的tetr蛋白,旨在四环素不存在的情况下更强烈地阻抑shrna表达。为此,我们进行了细菌性tetrcdna的多参数rna和密码子优化,并且使用所得到的密码子优化的tetr(opttetr)生成新的egfp可诱导敲低hesc系(图14b)。与标准序列(stdtetr;图14d)相比时,这种修饰允许tetr表达增加十倍。另外,opttetr纯合表达足以彻底防止shrna泄露,同时充分保留高效的敲低诱导(图14c)。值得注意地,可诱导敲低是快速、可逆和剂量响应的(图14e、f)。最后,诱导型hesc展示正常核型(数据未显示),表明为产生这些系所必需的基因组工程化并不改变它们的遗传稳定性。
基于这些令人鼓舞的结果,我们进一步在内源基因的背景下,通过生成携带针对pou5f1/oct4或b2m的诱导型shrna的hesc,验证了这种方法(数据未显示)。值得注意地,分析的全部子系(每种基因6个子系)均显示稳健的可诱导敲低,无明显的shrna泄露。四环素滴定法鉴定了部分或完全敲低oct4的最佳浓度。如预期,oct4强烈下降特别地导致多能性的丧失和神经外胚层标志物及定形内胚层标志物的诱导。用20个额外的oct4可诱导敲低hesc子系获得相似结果,从而确认这种方法的稳健性和重现性。重要地,具有强力和严密受调节的敲低的hesc的生成是如此高效,从而在混合的细胞群体上进行抗生素选择后,可以立即进行表型分析,因而完全绕过挑出单个集落供克隆性分离的必要。总之,这些结果确立了,用优化的可诱导敲低系统对gsh双重打靶是控制hpsc中基因表达的有力方法。这种方案此后命名为optikd,即,优化的可诱导敲低(图14a)。
实施例6
敲低多种已分化细胞中基因的能力将代表超过先前诱导型基因敲低系统的显著进展。为了彻底检验这种可能性,我们分析了optikd平台在分化成三种胚层的hpsc中以及在一组充分分化的十三个细胞类型中敲低egfp转基因的有效性(图15a)。对于两种方法,qpcr分析表明,测试的全部谱系中强烈和可诱导地敲低egfp转录物(图17)。显微观察确认egfp蛋白表达稳健下降,并且流式细胞术显示对于大部分谱系,egfp荧光下降超过70%(数据未显示)。
实施例7
在hpsc中开发优化的诱导型crispr/cas9敲除平台。
我们将我们的注意力转向开发可诱导敲除方案。当前的可诱导crispr/cas9方法依赖于cas9在组成型表达的grna存在下的条件性过量表达。在这种情况下,通过tet-on方法实现控制cas9过量表达,在所述方法中,多西环素处理后,四环素控制的逆向反式激活因子(rtta)激活polii-依赖性四环素应答元件(tre)启动子(多个tet操纵子和最小cmv启动子之间的融合物)。尽管这个tet-on平台已经成功地应用于某些人细胞类型,但是我们观察到,这种诱导型系统在hpsc分化成多个谱系(包括心肌细胞、肝细胞和平滑肌细胞)期间,甚至在靶向入aavs1gsh后被沉默(数据未显示)。我们探索了通过组成型表达的cag启动子驱动的cas9与grna可诱导盒组合,开发替代性和改进方法的可能性,其中所述可诱导盒基于一种为诱导型shrna表达所开发的可诱导盒(图18a、b)。我们因此生成了其中荧光报道基因可以按照可诱导方式敲除的hesc系(图18c)。为此,我们用诱导型egfpgrna和组成型cas9二者在aavs1基因座对rosa26-egfpd2报道hesc打靶,每个转基因整合入两个等位基因之一中。这个双重打靶方案是快速(<2周)和高效的(>90%的系含有两种转基因)。值得注意地,在四环素存在下培育独立的克隆子系时,我们观察到全部已打靶的系中egfpd2表达减少,并且在早至四环素诱导后五天,egfpd2纯合细胞显示近乎均匀丢失报道基因的至少一个拷贝(如egfpd2荧光减少50%所展示)。延长的四环素处理导致多达75%的egfpd2纯合细胞中egfpd2荧光逐渐完全丢失(数据未显示)。有趣地,从相同aavs1基因座共表达相同egfpgrna盒的两个或三个拷贝足以显著地增加所分析的全部克隆性子系中诱导型egfpd2敲除的速度和效率。例如,四环素处理后,同时诱导相同grna的三个拷贝产生引人注目的95%敲除效率。重要地,在四环素不存在的情况下,延长培养后,诱导型egfpd2敲除的hesc在egfpd2阳性细胞比例和它们的荧光方面均未显示任何显著减少,甚至当使用几个grna拷贝时也是如此。这显示诱导型grna表达紧密受控。最后,检验针对egfpd2的附加grna揭示,可诱导敲除的速度和效率强烈地依赖于grna。实际上,最佳序列允许在仅2天诱导后多达90%的敲除。值得注意地,最高效的grna还导致失控的egfpd2敲除,但是通过简单地添加第二tet操纵子至诱导型h1启动子以确保甚至更严格的转录性控制,避免了这种局限。总体上,这些结果显示,可以轻易地修改该敲低系统以支持诱导型grna表达并且在类型广泛的grna效力范围内允许紧密受控的crispr/cas9活性。尽我们所知,这是首个基于诱导型grna表达的条件性crispr/cas9方案。
- 还没有人留言评论。精彩留言会获得点赞!