本发明属于医药技术领域,具体涉及一种高纯度沙利度胺α晶型的制备方法。
背景技术:
沙利度胺thalidomide(distaval)是一种合成的谷氨酸衍生物,别名:沙立度胺,反应停,酞胺哌啶酮,酞胺哌酮、酞谷酰亚胺、酞咪哌啶酮,其结构式如下:
文献j.chem.soc.perkintrans.21994(2063-2067)对沙利度胺的晶型进行了研究,发现沙利度胺存在两种晶型,分别是α晶型和β晶型,用差示扫描量热法(dsc)检测发现,α晶型在271.5-273.0℃吸热峰,β晶型仅在275.0-276.5℃有吸热峰,研究认为α晶型在dsc检测过程中会部分或全部转成β晶型,从而又出现β晶型的吸热峰。α晶型的x-射线衍射图(xrpd)在约11.33、14.32、19.20、22.77、26.12、30.36处包含特征峰;β晶型的x-射线衍射图(xrpd)在约11.78、12.96、13.75、17.06、19.26、24.06、25.73、29.05、29.29处包含特征峰。
沙利度胺传统的结晶方法是以极性溶剂吡啶、二甲基亚砜、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷烃等为良性溶剂,以水、甲醇、乙醇、丙醇、丁醇、丙酮等溶剂为不良溶剂进行结晶。专利cn103068812a描述了用二甲基甲酰胺、二甲基乙酰胺做第一溶剂,丙酮、甲醇和水做第二溶剂重结晶制备α晶型沙利度胺。专利cn102924432a描述了用热的二甲基亚砜或热的二甲基亚砜与脂肪醇的混合溶剂溶解沙利度胺粗品,然后活性炭脱水、趁热过滤,滤液降温析晶的方法制备高纯度的沙利度胺,但是没有dsc和xrpd等晶型检测数据。专利cn102260241a描述了用极性溶剂溶剂,再加醇析晶制备的α晶型沙利度胺方法。以上方法都需要用到高沸点的极性溶剂,以及酮类、醇类等溶剂,结晶后的母液非常难于重复利用,社会成本和经济成本较高。
因此,有必要开发出更优的制备沙利度胺α晶型的方法,为该药物的后续工业化生产提供更好的方案。
技术实现要素:
本发明的目的在于提供一种成本低、操作简便、安全环保、可以循环套用、适合工业化大生产的制备沙利度胺α晶型的方法。
本发明的第一方面,提供了一种工业化制备沙利度胺α晶型的方法,所述方法包括步骤:
(1)提供第一溶液,所述第一溶液包含混合溶剂以及溶解于所述混合溶剂中的沙利度胺,其中,所述混合溶剂为四氢呋喃和水的混合溶剂;
(2)对所述第一溶液进行选择性蒸馏,从而获得第一馏出物和经蒸馏后的第一混合物,其中,所述第一混合物含有残留的混合溶剂、溶于所述混合溶剂的沙利度胺以及析出的沙利度胺α晶型,其中,所述的选择性蒸馏是使四氢呋喃被蒸馏出而水基本上不被蒸馏出;
(3)从所述第一混合物中,分离出所述的沙利度胺α晶型;和
(4)对所述分离的沙利度胺α晶型进行洗涤和干燥,从而获得经干燥的沙利度胺α晶型。
在另一优选例中,对于在步骤(2)获得的第一馏出物(以四氢呋喃为主),进行回收利用。
在另一优选例中,所述的经回收的第一馏出物用于制备所述的第一溶液。
在另一优选例中,在所述的第一馏出物中,四氢呋喃:水的重量比为≥10:1,较佳地≥20:1,更佳地≥50:1。
在另一优选例中,所述“水基本上不蒸出”指在所述的第一馏出物中,四氢呋喃:水的重量比为≥10:1,较佳地≥20:1,更佳地≥50:1。
在另一优选例中,在所述的第一混合物中,四氢呋喃:水的重量比为≤1/10,较佳地≤1/20,更佳地≤1/100。
在另一优选例中,在所述的第一溶液中,四氢呋喃:水的重量比为100:1-1:10,较佳地10:1-1:5,更佳地3:1-1:1。
在另一优选例中,所述混合溶剂还含有少量吡啶,较佳地吡啶和四氢呋喃的质量比为1:500-1:5,较佳地1:100-1:10。
在另一优选例中,所述沙利度胺与混合溶剂中四氢呋喃的质量比为1:0.5-300,较佳地1:1-100,更佳地1:1-50,更佳地1:3-30,更佳地1:1-10。
在另一优选例中,所述沙利度胺和混合溶剂中水的质量比为1:1-50,较佳地1:3-50,更佳地1:3-10,更佳地1:1-3。
在另一优选例中,所述方法还包括:位于步骤(1)之前的步骤(0):将沙利度胺粗品与水和四氢呋喃进行混合,从而获得第一溶液。
在另一优选例中,所述方法还包括:将回收的馏出物用于步骤(0)。
在另一优选例中,所述方法中,步骤(0)至步骤(2)是循环进行和连续进行的。
在另一优选例中,所述方法循环的次数为1-1000次,较佳地2-100次。
在另一优选例中,所述溶解为室温溶解或加热溶解。较佳地,所述室温为15-40℃;所述加热为40-100℃。
在另一优选例中,所述蒸馏在t1温度下进行,其中所述t1为15-100℃,较佳地为15-40℃或40-100℃。
在另一优选例中,所述蒸馏为减压蒸馏或常压蒸馏。
在另一优选例中,所述第一溶液的体积为1-5000l,较佳地10-500l。
在另一优选例中,所述分离为过滤或抽滤。
在另一优选例中,所述方法的收率在95%以上。
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
附图说明
图1显示了实施例1中制得的沙利度胺α晶型的dsc图谱。
图2a显示了实施例1中制得的沙利度胺α晶型的xrpd图谱;图2b显示了实施例1中制得的沙利度胺α晶型的x-射线衍射数据。
图3显示了实施例2中制得的沙利度胺α晶型的dsc图谱。
图4a显示了实施例2中制得的沙利度胺α晶型的xrpd图谱;图4b显示了实施例2中制得的沙利度胺α晶型的x-射线衍射数据。
图5显示了实施例3中制得的沙利度胺α晶型的dsc图谱。
图6a显示了实施例3中制得的沙利度胺α晶型的xrpd图谱;图6b显示了实施例3中制得的沙利度胺α晶型的x-射线衍射数据。
图7显示了实施例4中制得的沙利度胺α晶型的dsc图谱。
图8a显示了实施例4中制得的沙利度胺α晶型的xrpd图谱;图8b显示了实施例4中制得的沙利度胺α晶型的x-射线衍射数据。
图9显示了实施例5中制备沙利度胺α晶型的工艺流程图。
具体实施方式
本发明人通过广泛而深入的研究,首次研发出一种成本低、操作简便、安全环保、可以循环使用的制备沙利度胺α晶型的方法。本发明采用四氢呋喃和水的混合溶剂溶解沙利度胺,并进行蒸馏析晶得到沙利度胺α晶型。本发明制备方法中四氢呋喃可通过简单的蒸馏回收,易于重复利用,成本低,对环境绿色友好。采用低沸点的四氢呋喃,溶剂容易去除、残留少,制备工艺简单高效、条件温和、重复性好、成本低廉、收率高(≥97%),适合大规模工业化生产。通过本发明方法制备的晶体,纯度高、结晶度高、溶剂残留少。在此基础上,完成了本发明。
术语说明
除非另外定义,否则本文中所用的全部技术与科学术语均具有如本发明所属领域的普通技术人员通常理解的相同含义。
如本文所用,在提到具体列举的数值中使用时,术语“约”意指该值可以从列举的值变动不多于1%。例如,如本文所用,表述“约100”包括99和101和之间的全部值(例如,99.1、99.2、99.3、99.4等)。
如本文所用,术语“含有”或“包括(包含)”可以是开放式、半封闭式和封闭式的。换言之,所述术语也包括“基本上由…构成”、或“由…构成”。
沙利度胺
沙利度胺thalidomide(distaval)是一种合成的谷氨酸衍生物,别名:沙立度胺,反应停,酞胺哌啶酮,酞胺哌酮、酞谷酰亚胺、酞咪哌啶酮,其结构式如下:
20世纪50年代,由德国开发此药物主要是用于治疗癫痫,但由于缺乏有效性,随后被作为一种睡眠辅助用药,同时在怀孕期间广泛用于孕妇止吐。20世纪60年代初,反应停事件---出现大量的沙利度胺导致的婴儿畸形报道(如:短肢畸形、长骨缺损、耳廓缺失、唇裂、心脏和胃肠道畸形等),从而被很多国家禁止使用,并撤出医药市场,然而科学家并未全盘否定沙利度胺,继续对它进行深入研究,特别是在免疫、抗炎、抗血管生成的药理和一些疑难病症上的临床治疗研究中取得了令人欣喜和鼓舞的结果,从而使人们对沙利度胺又有了新的认识。20世纪70年代起,随着对麻风、风湿病和多种类型恶性肿瘤的研究进展陆续出现,以色列皮肤病学沙利度胺作为镇静药给麻风性结节红斑病人使用,症状迅速改善。以后许多例麻风性结节红斑病人使用反应均获得很好的治疗效果,1998年美国fda批准沙利度胺用于治疗麻风结节性红斑。2004年美国血液学学会年会上,美国梅奥医院rajkumar报告了沙利度胺及其类似物一线治疗多发性骨髓瘤的2项研究结果,沙利度胺及其类似物均可有效治疗多发性骨髓瘤。1998年,美国fda批准用于治疗麻风病伴随症。2006年5月美国fda批准用于治疗多发性骨髓瘤。2010年沙利度胺在中国被批准上市,除了可以治疗麻风结节性红斑以外,在临床诊疗指南血液学分册中沙利度胺可以治疗多发性骨髓瘤;在临床诊疗指南风湿学分册中沙利度胺可以治疗强直性脊柱炎和白塞氏症。
文献j.chem.soc.perkintrans.21994(2063-2067)对沙利度胺的晶型进行了研究,发现沙利度胺存在两种晶型,分别是α晶型和β晶型,用差示扫描量热法(dsc)检测发现,α晶型在271.5-273.0℃吸热峰,β晶型仅在275.0-276.5℃有吸热峰,研究认为α晶型在dsc检测过程中会部分或全部转成β晶型,从而又出现β晶型的吸热峰。α晶型的x-射线衍射图(xrpd)在约11.33、14.32、19.20、22.77、26.12、30.36处包含特征峰;β晶型的x-射线衍射图(xrpd)在约11.78、12.96、13.75、17.06、19.26、24.06、25.73、29.05、29.29处包含特征峰。
沙利度胺传统的结晶方法是以极性溶剂吡啶、二甲基亚砜、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷烃等为良性溶剂,以水、甲醇、乙醇、丙醇、丁醇、丙酮等溶剂为不良溶剂进行结晶。专利cn103068812a描述了用二甲基甲酰胺、二甲基乙酰胺做第一溶剂,丙酮、甲醇和水做第二溶剂重结晶制备α晶型沙利度胺。专利cn102924432a描述了用热的二甲基亚砜或热的二甲基亚砜与脂肪醇的混合溶剂溶解沙利度胺粗品,然后活性炭脱水、趁热过滤,滤液降温析晶的方法制备高纯度的沙利度胺,但是没有dsc和xrpd等晶型检测数据。专利cn102260241a描述了用极性溶剂溶剂,再加醇析晶制备的α晶型沙利度胺方法。以上方法都需要用到高沸点的极性溶剂,以及酮类、醇类等溶剂,结晶后的母液非常难于重复利用,社会成本和经济成本较高。
结晶
可以通过操作溶液,使得感兴趣化合物的溶解度极限被超过,从而完成生产规模的结晶。这可以通过多种方法来完成,例如,在相对高的温度下溶解化合物,然后冷却溶液至饱和极限以下。或者通过沸腾、常压蒸发、真空干燥或通过其它的一些方法来减小液体体积。可通过加入抗溶剂或化合物在其中具有低的溶解度的溶剂或这样的溶剂的混合物,来降低感兴趣化合物的溶解度。另一种可选方法是调节ph值以降低溶解度。有关结晶方面的详细描述请参见crystallization,第三版,jwmullens,butterworth-heinemanltd.,1993,isbn0750611294。
假如期望盐的形成与结晶同时发生,如果盐在反应介质中比原料溶解度小,那么加入适当的酸或碱可导致所需盐的直接结晶。同样,在最终想要的形式比反应物溶解度小的介质中,合成反应的完成可使最终产物直接结晶。
结晶的优化可包括用所需形式的晶体作为晶种接种于结晶介质中。另外,许多结晶方法使用上述策略的组合。比如在高温下将感兴趣的化合物溶解在溶剂中,随后通过受控方式加入适当体积的抗溶剂,以使体系正好在饱和水平之下。此时,可加入所需形式的晶种(并保持晶种的完整性),将体系冷却以完成结晶。
在本发明的一个优选例中,采用四氢呋喃和水的混合溶剂溶解沙利度胺,并进行蒸馏析晶处理得到高纯度的α晶型沙利度胺。
溶剂合物
化合物或药物分子与溶剂分子接触过程中,外部条件与内部条件因素造成溶剂分子与化合物分子形成共晶而残留在固体物质中的情况难以避免。化合物与溶剂结晶后形成的物质称作溶剂合物(solvate)。容易与有机化合物形成溶剂合物的溶剂种类为水、甲醇、苯、乙醇、醚、芳烃、杂环芳烃等。
水合物是一种特殊的溶剂合物。在制药工业中,无论在原料药的合成、药物制剂、药物贮存和药物活性评价中,水合物都因为其特殊性而具有单独讨论的价值。
示差扫描量热分析
又称“差示量热扫描分析”(dsc),是在加热过程中,测量被测物质与参比物之间的能量差与温度之间关系的一种技术。dsc图谱上的峰位置、形状和峰数目与物质的性质有关,故可以定性地用来鉴定物质。本领域常用该方法来检测物质的相变温度、玻璃化转变温度、反应热等多种参数。
在本发明中,差示扫描量热法(dsc)检测时开始以每分钟升10℃,从30℃升温到260℃,再以每分钟升1℃从260℃升温到285℃。
x-射线粉末衍射(xrd)
由包括以下的多种因素产生与这类x射线粉末衍射分析结果相关的测量差异:(a)样品制备物(例如样品高度)中的误差,(b)仪器误差,(c)校准差异,(d)操作人员误差(包括在测定峰位置时出现的误差),和(e)物质的性质(例如优选的定向误差)。校准误差和样品高度误差经常导致所有峰在相同方向中的位移。当使用平的支架时,样品高度的小差异将导致xrd峰位置的大位移。系统研究显示1mm的样品高度差异可以导致高至1°的2θ的峰位移。可以从x射线衍射图鉴定这些位移,并且可以通过针对所述位移进行补偿(将系统校准因子用于所有峰位置值)或再校准仪器消除所述位移。如上所述,通过应用系统校准因子使峰位置一致,可校正来自不同仪器的测量误差。本发明实施例中x-射线衍射是以丙硫菌唑溶剂化物为测定对象。
仪器型号:brukerd8advance,靶:cu靶,探测器:lynxeye,发散狭缝1.0mm,索拉狭缝0.4°,连续扫描,步长0.02°,速度8°/min,扫描范围为3-45°。
制备方法
本发明使用如本发明第一方面所述的方法制备沙利度胺晶体,所述方法包括步骤:
(1)提供第一溶液,所述第一溶液包含混合溶剂以及溶解于所述混合溶剂中的沙利度胺,其中,所述混合溶剂为四氢呋喃和水的混合溶剂;
(2)对所述第一溶液进行选择性蒸馏,从而获得第一馏出物和经蒸馏后的第一混合物,其中,所述第一混合物含有残留的混合溶剂、溶于所述混合溶剂的沙利度胺以及析出的沙利度胺α晶型,其中,所述的选择性蒸馏是使四氢呋喃被蒸馏出而水基本上不被蒸馏出;
(3)从所述第一混合物中,分离出所述的沙利度胺α晶型;和
(4)对所述分离的沙利度胺α晶型进行洗涤和干燥,从而获得经干燥的沙利度胺α晶型。
在本发明的一个优选实施方式中,把沙利度胺粗品、四氢呋喃、水分别加入结晶釜,加热搅拌溶解,直到完全溶清,然后蒸馏回收四氢呋喃,待四氢呋喃基本完全回收,冷却到室温,过滤,滤饼用适量的水洗涤三次,滤饼烘干得到沙利度胺α晶型成品。
研究中发现,沙利度胺在四氢呋喃中极微溶解,在水中极微溶解,通常会认为他们都是沙利度胺的不良溶剂。但是出乎意料的发现:沙利度胺可以溶解在四氢呋喃和水的混合溶剂中,然后对比四氢呋喃和水的沸点差异,因此尝试用四氢呋喃和水的混合溶剂进行重结晶,然后回收四氢呋喃,让沙利度胺在水中析出,能够得到α晶型的沙利度胺。水的用量为沙利度胺粗品的3倍重量或以上,四氢呋喃的用量为粗品沙利度胺1倍量-300倍量,优选1倍量-50倍量,优选1倍量-10倍量,更满足结晶过滤工业化生产的物料转移方便的需求。
本发明采用四氢呋喃和水的混合溶剂溶解沙利度胺,并进行析晶处理(如蒸馏回收四氢呋喃)得到沙利度胺α晶型。本发明的制备方法采用低沸点的四氢呋喃,溶剂容易去除、残留少,制备工艺简单高效、条件温和、重复性好、成本低廉、收率高(≥95%),适合大规模工业化生产。通过本发明方法制备的晶体,纯度高、结晶度高、溶剂残留少。本发明方法制备过程中,溶剂循环使用,因此在制备等量的沙利度胺晶体时,溶剂使用量非常少,节省成本。
本发明的主要优点在于:
(1)本发明采用低沸点的混合溶剂,溶剂容易去除、残留少,本发明的制备方法操作简便,并可在较低温度下蒸馏析晶,耗能少,杂质少(制备的α晶型纯度高),工艺条件温和、简单高效、重复性好,溶剂可重复使用,成本低廉,工艺稳定,结晶收率在95%以上,耗时少,非常适合大规模生产。
(2)本发明方法不需要用到高沸点的极性溶剂和酮类、醇类等溶剂,所用的四氢呋喃溶剂可以通过简单的蒸馏回收,非常易于重复利用,并且即使回收的四氢呋喃中含有少量水,也可以直接重复利用(无需分离或去除其中的少量水),对环境绿色友好,并且制备方法简单高效,具有较高的经济和社会价值。
(3)本发明方法制备的α晶型纯度高,并且溶剂残留少,容易过滤洗涤,能很好地满足后续制剂对原料性质的需求。
(4)本发明方法制备过程中不需要加入晶种,一边回收溶剂一边析晶,操作简单,容易工业化。
(5)本发明方法中溶剂循环使用,因此在制备等量的沙利度胺晶体时,溶剂用量非常少,节省成本。并且采用特定的溶剂用量(如水的用量为沙利度胺粗品的3倍重量或以上,四氢呋喃的用量为沙利度胺粗品1倍量-50倍量),以及混合溶剂中特定的四氢呋喃和水的重量比,更满足结晶、过滤工业化生产的物料转移方便的需求。
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。
除非另行定义,文中所使用的所有专业与科学用语与本领域熟练人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法及材料皆可应用于本发明方法中。文中所述的较佳实施方法与材料仅作示范之用。以下实施例中所用的实验材料和试剂如无特别说明均可从市售渠道获得。常温或室温指4℃-40℃,较佳地15-30℃。
实施例1
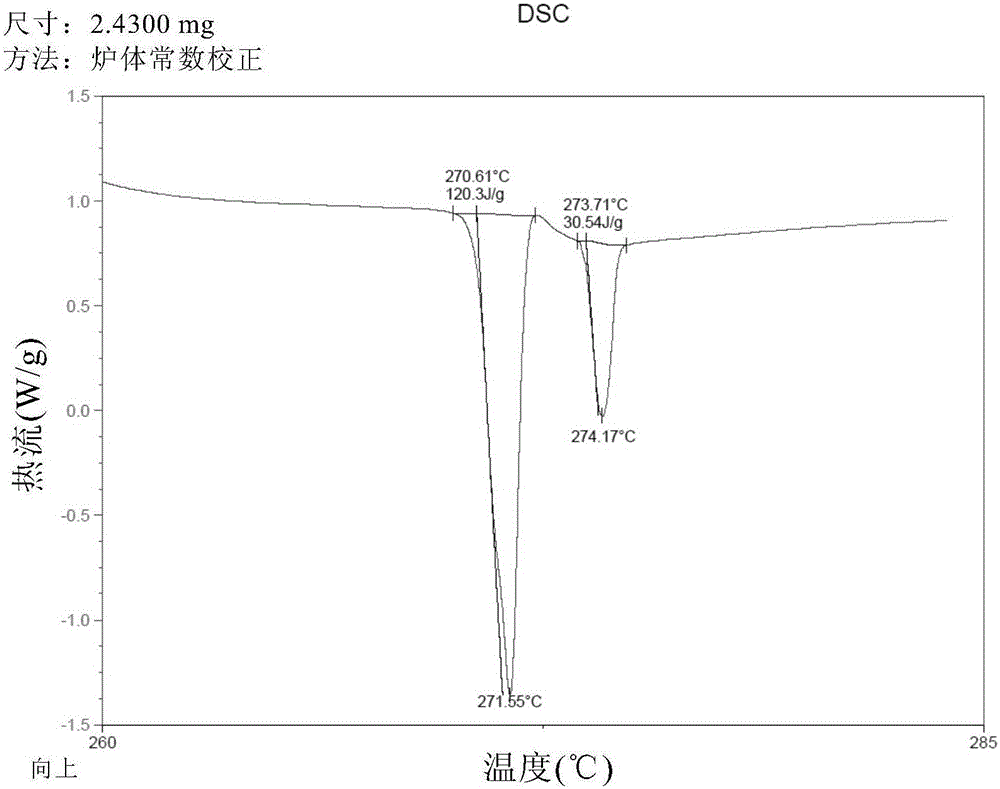
将沙利度胺粗品50g,四氢呋喃1500g先后加入2l的三口圆底烧瓶中,机械搅拌,油浴加热至回流,加入纯化水150g,固体全部溶解,保持油浴温度95-100℃,常压蒸馏四氢呋喃,四氢呋喃回收完毕,将母液冷却到室温,减压抽滤,滤饼再80℃真空干燥烘料4h,得到沙利度胺晶体成品48.5g,重结晶收率97.0%,纯度99.8%,四氢呋喃残留量几乎为0%。dsc检测结果如图1所示,dsc检测吸热峰为271.55℃(α晶型特征吸热峰)和274.17℃,表明所得的沙利度胺晶体是α晶型,并且纯度高。x-射线衍射图(xrpd)如图2a所示,x-射线衍射数据如图2b所示。结果表明得到的产品是α晶型的沙利度胺,并且纯度高。
实施例2
将沙利度胺粗品50g,四氢呋喃150g先后加入500ml的三口圆底烧瓶中,机械搅拌,油浴加热至回流,加入纯化水50g,固体全部溶解,降至室温减压浓缩回收四氢呋喃,四氢呋喃回收完毕,将母液抽滤,滤饼再80℃真空干燥烘料4h,得到沙利度胺晶体成品49.0g,重结晶收率98.0%,纯度99.7%,四氢呋喃残留量几乎为0%。dsc检测结果如图3所示,dsc检测吸热峰为271.47℃和274.32℃,表明所得的沙利度胺晶体是α晶型,并且纯度高。x-射线衍射图(xrpd)如图4a所示,x-射线衍射数据如图4b所示。也表明得到的产品是α晶型的沙利度胺,并且纯度高。
实施例3
将沙利度胺粗品50g,将实施例1中回收得到的四氢呋喃称取1500g先后加入2l的三口圆底烧瓶中,机械搅拌,油浴加热至回流,加入纯化水150g,固体全部溶解,降至室温减压浓缩回收四氢呋喃,四氢呋喃回收完毕,将母液抽滤,滤饼再80℃真空干燥烘料4h,得到沙利度胺晶体成品48.8g,重结晶收率97.6%,纯度99.8%,四氢呋喃残留量几乎为0%。dsc检测结果如图5所示,dsc检测吸热峰为271.31℃(α晶型特征吸热峰)和274.23℃,表明沙利度胺是α晶型,并且纯度高。x-射线衍射图(xrpd)如图6a所示,x-射线衍射数据如图6b所示。结果表明得到的产品是α晶型的沙利度胺,并且纯度高。
实施例4
将沙利度胺粗品50g,将实施例2中回收得到的四氢呋喃称取150g先后加入500ml的三口圆底烧瓶中,机械搅拌,油浴加热至回流,加入纯化水150g,固体全部溶解,降至室温减压浓缩回收四氢呋喃,四氢呋喃回收完毕,将母液抽滤,滤饼再80℃真空干燥烘料4h,得到沙利度胺晶体成品49.0g,重结晶收率98.0%,纯度99.7%,四氢呋喃残留量几乎为0%。dsc检测结果如图7所示,dsc检测吸热峰为271.32℃,表明所得的沙利度胺晶体是α晶型,并且纯度高。x-射线衍射图(xrpd)如图8a所示,x-射线衍射数据如图8b所示。结果表明得到的产品是α晶型的沙利度胺,并且纯度高。
实施例5
如图9所示,制备沙利度胺α晶型的步骤如下:
(1)将沙利度胺粗品50kg,四氢呋喃150kg,水150kg先后加入500l的结晶釜中,加热搅拌溶解,直至完全溶清,得到第一溶液;
(2)将第一溶液进行常压选择性蒸馏,使四氢呋喃被蒸馏出而水基本上不被蒸馏出,获得第一馏出物和经蒸馏后的第一混合物,所述第一混合物含有残留的混合溶剂、溶于所述混合溶剂的沙利度胺以及析出的沙利度胺结晶物;
(3)将经蒸馏后的第一混合物冷却到室温,减压抽滤;
(4)滤饼用适量的水洗涤三次并烘干,得到沙利度胺α晶型成品48kg;
(5)将步骤(2)获得的第一馏出物(以四氢呋喃为主)回收,加入到结晶釜中,并加入沙利度胺粗品50kg和水150kg,加热搅拌溶解,直至完全溶清,得到第一溶液;
(6)重复步骤(2)-(5)3次。
结果表明,使用150kg四氢呋喃和200kg沙利度胺粗品,即可制备得到192kg沙利度胺α晶型成品,相当于制备1kg高纯度的沙利度胺所需四氢呋喃约0.78kg,重结晶收率96%,纯度99.8%,四氢呋喃残留量几乎为0%。dsc检测吸热峰与图1相似,在α晶型特征峰处有吸热峰,表明制备得到的是高纯度的α晶型的沙利度胺。由于四氢呋喃是重复利用4次,因此,共使用150kg四氢呋喃和200kg沙利度胺粗品,即得到192kgα晶型成品。
对比例1
将沙利度胺粗品50g,二甲基亚砜100g先后加入1l的三口圆底烧瓶中,机械搅拌,油浴加热至90℃,然后保温搅拌直到溶清,然后降温到20-40℃后慢慢加入乙醇250g,抽滤,滤饼用少量乙醇洗涤,滤饼转移到80℃真空干燥中烘料4h,得沙利度胺成品35.0g,重结晶收率70.0%,dsc检测吸热峰在274.30℃附近有吸热峰,在271.30℃附近没有吸热峰(α晶型的特征吸热峰),这表明得到的沙利度胺成品中含有大量的β晶型。并且最后结晶后的母液为二甲基亚砜和乙醇的混合物,由于二甲基亚砜为溶剂,而乙醇为反溶剂,因此不能直接重复利用,需要进行分离。
对比例2
将沙利度胺粗品50g,二甲基亚砜100g先后加入1l的三口圆底烧瓶中,机械搅拌,油浴加热至溶清,加入乙醇300g,冷却到室温,减压抽滤,滤饼再80℃真空干燥烘料4h,得到沙利度胺成品38g。dsc检测,没有发现α晶型的吸热峰。
对比例3
将沙利度胺粗品50g,二甲基亚砜100g先后加入1l的三口圆底烧瓶中,机械搅拌,油浴加热至90℃,然后保温搅拌直到溶清,然后降温到20-40℃后慢慢加入纯化水250g,抽滤,滤饼用少量纯化水洗涤,滤饼转移到80℃真空干燥中烘料4h,得沙利度胺成品45.0g,重结晶收率90.0%,dsc检测在274.30℃附近有吸热峰,在271.30℃附近没有吸热峰(α晶型的特征吸热峰)。这表明得到的沙利度胺成品中含有大量的β晶型。并且最后结晶后的母液为二甲基亚砜和水的混合物,由于二甲基亚砜为溶剂,而水为反溶剂,因此不能直接重复利用,需要进行分离。
对比例4
将沙利度胺粗品50g,吡啶150g先后加入1l的三口圆底烧瓶中,机械搅拌,油浴加热至回流溶清,加入纯化水300g,冷却到室温,减压抽滤,滤饼再80℃真空干燥烘料4h,得α晶型的沙利度胺成品40g。沙利度胺成品中吡啶残留容易超标,并且吡啶废水很难处理。
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。