水性含氟丙烯酸酯共聚物、杂化膜及其制备方法和应用与流程
本发明涉及一种水性含氟丙烯酸酯共聚物、杂化膜及其制备方法和应用。
背景技术:
含氟聚合物由于碳氟键极强的稳定性,含氟聚合物具有很好的化学稳定性、热稳定性、优异的表面性能、优异的润滑性、良好的电绝缘性、耐老化性、抗辐射性和极小的吸水性等优良的综合性能,在织物整理、功能涂层、航空航天、生物医药以及微电子等领域已得到广泛的应用。含氟丙烯酸酯共聚物是一类由含氟丙烯酸酯单体与其他功能单体共聚而得的含氟聚合物,其通常具有高拒水性、高成膜性、高粘附力等。
近年来,随着人们环保意识的加强,环境友好的含氟丙烯酸聚合物乳液逐渐引起重视。利用含氟化合物的优越性能制备用于织物的拒水拒油整理剂的专利文献多有报道。专利us6126849、us6451717、us5344903分别公开了一种将含氟丙烯酸酯混合单体、丙烯酸十八酯、n-羟基丙烯酰胺与含氟乳化剂混合经高压均质乳化,v-50引发,制备转化率在99%以上的乳液,具有很好的拒水拒油性能。us5055538公开了一种将全氟辛基乙基丙烯酸酯与丙烯酸十八酯、丙烯酸乙酯、n-羟基丙烯酰胺、1,2-环氧乙烷乙烯基丙烯酸甲酯、含氟乳化剂混合乳化,v-50引发制备含氟聚丙烯酸酯乳液的方法,所得乳液拒水拒油性良好,耐水洗,可与其他助剂复配。cn1493601公开了一种水乳型拒水拒油剂及其制备方法,cn201010520150.7、cn200510038650.6等分别合成了性能各异的含氟聚丙烯酸酯乳液。上述的含氟聚丙烯酸酯乳液方法中存在的问题主要有两个方面:一是制备的含氟聚丙烯酸酯乳液大部分全氟侧链含氟碳原子数≥8,具有生物累积性和生物毒性,有关法案已禁止使用;二是使用的引发剂、乳化剂成本较高,不利于大规模推广应用,乳化过程中加入如丙酮等助乳化剂,具有毒性,环境不友好;三是含氟聚丙烯酸酯结构单一,成本较高,同样限制了其推广应用。
由此,亟需开发一种既保证共聚物具有拒水拒污性能,又能降低含氟单体的用量,且碳氟原子小于8的含氟聚丙烯酸酯共聚物。
技术实现要素:
本发明所要解决的技术问题是提供一种不同于现有技术的水性含氟丙烯酸酯共聚物、杂化膜及其制备方法和应用。本发明的水性含氟丙烯酸酯共聚物中含氟丙烯酸酯中含氟碳原子数≤6,用量少,由该水性含氟丙烯酸酯共聚物制得的杂化膜在拒水拒污、成膜均匀和防酸防碱的防腐方面效果好,可用于油水分离,且该制备方法仅以水为溶剂,绿色环保,成本低,简单高效,具有较好的市场应用前景。
本发明通过下述技术方案来解决上述技术问题。
本发明提供了一种核壳型共聚物,所述核壳型共聚物以烷基丙烯酸酯单体聚合为核,以烷基丙烯酸酯单体、含氟丙烯酸酯单体和丙烯酸羟乙酯单体共聚为壳;
其中,所述烷基丙烯酸酯单体质量占总单体质量的质量百分比为78.13%~92.70%,所述含氟丙烯酸酯单体质量占总单体质量的质量百分比为6.21~20.94%,所述丙烯酸羟乙酯单体质量占总单体质量的质量百分比为0.91%~1.09%;
所述烷基丙烯酸酯单体结构如式1所示,所述含氟丙烯酸酯单体结构如式2所示,所述丙烯酸羟乙酯单体结构如式3所示:
r1为c1-c18烷基;r2和r3各自独立地为h或ch3;rf为cnf2n+1ch2ch2-,n选自2~6中任意整数。
其中,所述烷基丙烯酸酯单体质量,所述含氟丙烯酸酯单体质量,所述丙烯酸羟乙酯单体质量均为聚合为核或壳之前的质量;所述总单体质量为所述烷基丙烯酸酯单体,所述含氟丙烯酸酯单体和所述丙烯酸羟乙酯在聚合为核或壳之前的总质量。
本发明中,所述烷基丙烯酸酯单体质量占总单体质量的质量百分比可为78.13%~87.27%,所述含氟丙烯酸酯单体质量占总单体质量的质量百分比可为11.69~20.94%,所述丙烯酸羟乙酯单体质量占总单体质量的质量百分比可为0.91%~1.02%。
本发明中,所述rf中cnf2n+1可为直链结构或支链结构;较佳地rf中cnf2n+1为支链结构,n为5或6,例如:
本发明中,所述含氟丙烯酸酯单体可为2-(全氟己基)乙基甲基丙烯酸酯、2-(全氟己基)乙基丙烯酸酯和2-(全氟1,1-二甲基-丁基)乙基丙烯酸酯中的一种或多种,较佳地为2-(全氟己基)乙基丙烯酸酯和/或2-(全氟1,1-二甲基-丁基)乙基丙烯酸酯,例如2-(全氟1,1-二甲基-丁基)乙基丙烯酸酯。
本发明中,所述烷基丙烯酸酯单体可为本领域常规烷基丙烯酸酯单体,较佳地为甲基丙烯酸甲酯、丙烯酸甲酯、甲基丙烯酸丁酯、丙烯酸丁酯、甲基丙烯酸异辛酯、丙烯酸异辛酯、甲基丙烯酸月桂酯、丙烯酸月桂酯、甲基丙烯酸十八酯和丙烯酸十八酯中的一种或多种;更佳地为甲基丙烯酸甲酯、甲基丙烯酸丁酯和丙烯酸十八酯中的一种或多种。
本发明还提供了一种核壳型共聚物的制备方法,包括下述步骤:
步骤1、在引发剂条件下,将所述烷基丙烯酸酯单体和乳化剂与溶剂混合后得到的预乳液i进行聚合反应,即得核壳型共聚物的核乳液;
步骤2、在引发剂条件下,将所述烷基丙烯酸酯单体、所述含氟丙烯酸酯单体、所述丙烯酸羟乙酯单体和乳化剂与溶剂混合得到的预乳液ii,与所述步骤1中所得的核壳型共聚物的核乳液进行聚合反应,即得核壳型共聚物乳液;
步骤1中,所述烷基丙烯酸酯单体质量与总单体质量的质量百分比为40.84~48.46%;
步骤2中,所述烷基丙烯酸酯单体质量与总单体质量的质量百分比为37.28~44.23%;所述含氟丙烯酸酯单体质量与总单体质量的质量百分比为6.21~20.95%;所述丙烯酸羟乙酯单体质量与总单体质量的质量百分比为0.92~1.08%。
本发明中,所述核壳型共聚物的制备方法可为本领域中此类反应常规的制备方法,所述核壳型共聚物的制备方法的参数和条件可参照本领域此类反应常规的参数和条件。
其中,所述步骤1包括,在惰性气体保护下,将所述烷基丙烯酸酯单体、乳化剂与溶剂混合,得预乳液i,在引发剂下,将所述烷基丙烯酸酯单体进行聚合反应;较佳地,将所述预乳液i分批加入反应液中进行聚合反应;更佳地,将1/3所述预乳液i进行聚合反应,反应液呈蓝相后,滴加2/3所述预乳液i至反应液中进行聚合反应。
其中,所述步骤2包括,在惰性气体保护下,将所述烷基丙烯酸酯单体、所述含氟丙烯酸酯单体、所述丙烯酸羟乙酯单体和乳化剂与溶剂混合,得预乳液ii,在引发剂下,所述预乳液ii与所述步骤1所得的核壳型共聚物的核乳液进行聚合反应;较佳地为,将引发剂和所述预乳液ii滴加至所述步骤1所得的核结构乳液进行聚合。
步骤1中,所述烷基丙烯酸酯单体质量与所述总单体质量的质量百分比可为40.84~45.46%;
步骤2中,所述烷基丙烯酸酯单体质量与所述总单体质量的质量百分比可为37.28~41.64%;所述含氟丙烯酸酯单体质量与所述总单体质量的质量百分比可为11.69~20.94%;所述丙烯酸羟乙酯单体质量与所述总单体质量的质量百分比可为0.92~1.02%。
其中,所述溶剂可为本领域中此类反应常规溶剂,较佳地为去离子水。
其中,所述溶剂总质量与所述总单体质量的比值可为1~5,较佳地为1.8~2.3,更佳地为2.0~2.3。
其中,所述乳化剂可为复合乳化剂或反应型单一乳化剂;较佳地为反应型单一乳化剂;更佳地为1-烯丙氧基-3-(4-壬基苯酚)-2-丙醇聚氧乙烯(10)醚硫酸铵(dns-86)。
所述的复合乳化剂可为离子表面活性剂和非离子表面活性剂质量的组合物,较佳地为离子表面活性剂的质量与非离子表面活性剂的质量比为1:1~2.5。
所述的阳离子表面活性剂可为十二烷基三甲基氯化铵、十六烷基三甲基溴化铵和十八烷基三甲基氯化铵中的一种或多种;较佳地为,十二烷基三甲基氯化铵、十六烷基三甲基溴化铵和十八烷基三甲基氯化铵中的一种。
所述阴离子表面活性剂可为十二烷基硫酸钠、十二烷基磺酸钠和十二烷基苯磺酸钠中的一种或多种;较佳地为十二烷基硫酸钠、十二烷基磺酸钠和十二烷基苯磺酸钠中的一种。
所述非离子表面活性剂可为op(辛基酚聚氧乙烯醚)系列表面活性剂、np(壬基酚聚氧乙烯醚)系列表面活性剂、吐温系列表面活性剂或司班系列表面活性剂中的一种或多种;较佳地为op(辛基酚聚氧乙烯醚)系列表面活性剂、np(壬基酚聚氧乙烯醚)系列表面活性剂、吐温系列表面活性剂或司班系列表面活性剂中的一种。
其中,所述乳化剂的总质量与总单体质量的质量百分比可为2.1~2.4%,较佳地为2.1~2.3%。
其中,所述引发剂可为本领域中此类反应常规引发剂,较佳地为过硫酸铵、过硫酸钾和过硫酸钠中一种或多种;更佳地为过硫酸铵。
其中,所述引发剂总质量与所述总单体质量的质量百分比可为0.62~2.09%,较佳地为1.17~2.09%。
其中,所述引发剂的浓度的质量百分比可为本领域中此类反应常规浓度,较佳地为1.0~1.5%。
其中,步骤1中,所述引发聚合反应的温度可为本领域中此类反应常规温度,较佳地为40~80℃,更佳地为50~60℃。
其中,步骤1中,所述引发聚合反应的时间可为本领域中此类反应常规时间,较佳地为2~6小时,更佳地为6小时。
其中,步骤2中,所述聚合反应的温度可为本领域中此类反应常规温度,较佳地70~80℃。
其中,步骤2中,所述聚合反应的时间可为本领域中此类反应常规时间,较佳地为2~6小时。
其中,所述核壳型共聚物的制备方法在所述步骤2结束后,较佳地还包括将所述核壳型共聚物的乳液的ph调至7~9即可;更佳地为将所述核壳型共聚物的乳液降温至40℃,ph调至7~9。
其中,所述调节反应液的ph的试剂为本领域中此类反应常规试剂,较佳地为氨水。
所述烷基丙烯酸酯单体质量,所述含氟丙烯酸酯单体质量,所述丙烯酸羟乙酯单体质量均为聚合为核或壳之前的质量;所述总单体质量为所述烷基丙烯酸酯单体,所述含氟丙烯酸酯单体和所述丙烯酸羟乙酯在聚合为核或壳之前的总质量;所述溶剂总质量为步骤1和步骤2中溶剂的总质量;所述引发剂总质量为步骤1和步骤2中引发剂的总质量;所述乳化剂总质量为步骤1和步骤2中乳化剂的总质量。
本发明还提供了一种由上述制备方法制得的核壳型共聚物。
本发明还提供了所述核壳型共聚物在拒水拒污中的应用。
本发明还提供了一种拒水拒污整理剂,包含所述核壳型共聚物。
本发明还提供了一种杂化膜,包含所述核壳型共聚物、无机亲水性纳米材料和水性固化剂组分。
其中,所述无机亲水性纳米材料可为本领域的常规材料,较佳地为亲水性纳米二氧化硅、亲水性纳米二氧化钛或亲水性纳米氧化锌,更佳地为亲水性纳米二氧化硅。所述无机亲水性纳米材料粒径较佳地为10~50nm,更佳地为15~20nm。
其中,所述水性固化剂可为本领域常规水性固化剂,较佳地为水性异氰酸酯固化剂或水性氨基固化剂,例如水性异氰酸酯固化剂。
本发明还提供了一种所述杂化膜的制备方法,包括以下步骤:
将杂化膜液涂至基材上,成膜后,将基材固化即得;其中,所述杂化膜液为含有所述核壳型共聚物、无机亲水性纳米材料、水性固化剂和去离子水;较佳地为,将质量百分比为10%无机亲水性纳米材料水溶液,与核壳型共聚物、水性固化剂和去离子水混合,即可。
其中,所述固化的操作和条件可为本领域常规的操作和条件,较佳地为80~100℃条件下固化0.5~3h。
其中,所述基材可为本领域的常规基材,较佳地为玻璃、织物、纸张、尼龙和金属中的一种。
本发明中,所述乳液、所述去离子水、所述水性固化剂和所述无机亲水性纳米材料的质量比可为本领域常规质量比,较佳地10~35:50~80:0.01~1:1~10,例如30:70:0.036:4。
其中,所述涂布方法可为本领域常规涂布方法,较佳地为喷笔涂布。
其中,所述喷笔涂布的操作和条件可为本领域常规操作和条件,较佳地为,喷笔涂布的条件为笔口径2~5mm,喷笔口与基底材料水平距离为10~30cm,压力为100~500kpa。
其中,所述混合的操作和条件可为本领域常规的操作和条件,较佳地为在超声下混合10~30min。
本发明还提供所述杂化膜在油水分离中的应用。
本发明所述核壳型共聚物的制备方法较佳地为:
步骤1、去离子水1.5g,甲基丙烯酸甲酯0.328g,甲基丙烯酸丁酯1.232g和乳化剂dns-860.04g,室温下剧烈搅拌0.5小时形成预乳化液ⅰ;加入去离子水1.5g,甲基丙烯酸甲酯0.396g,甲基丙烯酸丁酯0.528g,丙烯酸十八酯0.5g,丙烯酸羟乙酯0.035g,含氟丙烯酸酯单体0.4~0.8g和乳化剂dns-860.04g,室温下剧烈搅拌0.5小时形成预乳液ⅱ。去离子水3g与过硫酸铵0.04g在室温下溶解得过硫酸铵水溶液。
步骤2、将0.04gdns-86,在氮气保护下加入1/3引发剂水溶液、1/3预乳化液ⅰ和1.5ml蒸馏水,置于60℃下以250rpm转速搅拌20分钟,体系引发成功,出现蓝相。逐渐升温至70℃,逐滴缓慢加入剩余的2/3预乳化液ⅰ和1/3引发剂水溶液,滴加时间为90分钟。滴加完毕后,体系继续搅拌反应2小时。将预乳化液ⅱ与剩余的1/3aps水溶液缓慢滴加入反应体系中,滴加时间控制在2小时。滴加完毕后,体系继续搅拌反应2小时。降温至40℃,氨水调节体系ph为7~9,即可。
本发明所述杂化膜的制备方法较佳地为:
将粒径为15±5nm无机亲水纳米二氧化硅配制成浓度为质量百分比为10%水溶液。将上述所得乳液0.3g、去离子水0.7g、质量百分比为12%水性固化剂0.03g、质量百分比为10%亲水纳米二氧化硅水溶液0.4g,超声10-30min混合均匀,即可。将乳液置于喷笔中,喷笔口径2mm,喷笔口与基底材料水平距离为20cm,压力为200kpa,喷涂至基材上。基材置于80-100℃烘箱中固化1小时,即得杂化涂膜材料。
在不违背本领域常识的基础上,上述各较佳地条件,可任意组合,即得本发明各较佳实例。
本发明所用原料2-(全氟1,1-二甲基-丁基)乙基丙烯酸酯为自制,其它试剂和原料均市售可得。
本发明的积极进步效果在于:
1、本发明水性含氟聚丙烯酸酯共聚物,全氟侧链(rf)含氟碳原子数小于等于6,易降解,不具有生物毒性;
2、本发明水性含氟聚丙烯酸酯共聚物,含氟单体仅在壳层聚合,保障拒水拒污性的同时降低了含氟原料的使用量;
3、本发明水性含氟聚丙烯酸酯共聚物,不会改变基材本身具有的特性;
4、本发明水性含氟聚丙烯酸酯共聚物,具有防酸和防碱的防腐性能;
5、本发明水性含氟聚丙烯酸酯共聚物及其涂膜的制备方法中,均以水为溶剂,绿色环保,经济高效。
附图说明
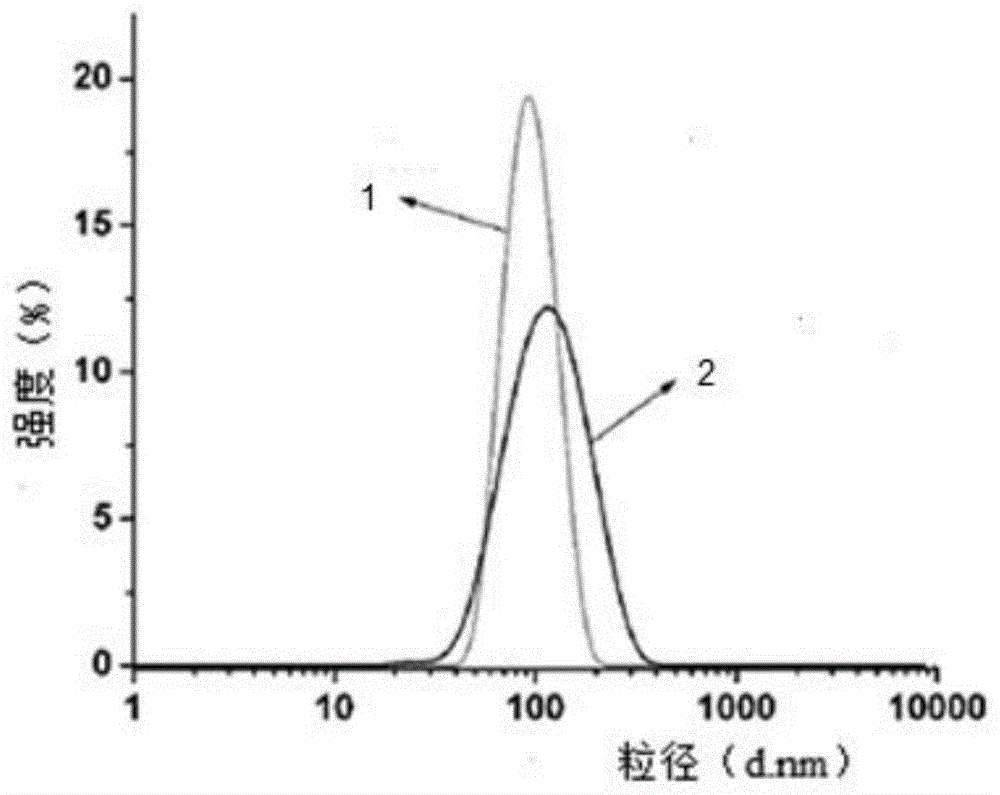
图1为实施例3核-壳结构乳胶粒粒径分布图,其中,1为核乳液粒子分布曲线,2为壳乳液粒子分布曲线。
图2为实施例3核-壳结构乳胶粒透射电镜图,其中(a)为未经染色核乳液,(b)经染色核乳液,(c)未经染色核-壳乳液,(d)经染色核-壳乳液。
图3为实施例6中棉布基材上杂化膜与水接触角图。
图4为实施例7杂化膜与水接触角图,其中,a为基底材料为棉布的杂化膜与水的接触角,b为基底材料为钢网(400目)的杂化膜与水的接触角,c为基底材料为尼龙网(400目)的杂化膜与水的接触角,d为基底材料为纸张的杂化膜与水的接触角。
图5为实施例7中杂化膜扫描电镜图,其中,a1为以玻璃为基材涂覆核-壳聚合物乳液的电镜图(标尺为50μm),a2为以玻璃为基材涂覆核-壳聚合物乳液的电镜图(标尺为10μm),a3为以玻璃为基材涂覆杂化膜液的电镜图(标尺为50μm),a4为以玻璃为基材涂覆杂化膜液的电镜图(标尺为10μm);
b1为以棉布为基材的电镜图(标尺为400μm),b2为以棉布为基材的电镜图(标尺为100μm),b3为以棉布为基材涂覆杂化膜液的电镜图(标尺为400μm),b4为以棉布为基材涂覆杂化膜液的电镜图(标尺为100μm);
c1为钢网为基材的电镜图(标尺为400μm),c2为钢网基材的电镜图(标尺为100μm),c3为以钢网为基材涂覆杂化膜液的电镜图(标尺为400μm),c4为以钢网为基材涂覆杂化膜液的电镜图(标尺为100μm);
d1为尼龙网基材的电镜图(标尺为400μm),d2为尼龙网基材的电镜图(标尺为100μm),d3为以尼龙网为基材涂覆杂化膜液的电镜图(标尺为400μm),d4为以尼龙网为基材涂覆杂化膜液的电镜图(标尺为100μm);
e1为纸为基材的电镜图(标尺为400μm),e2为纸为基材的电镜图(标尺为100μm),e3为以纸为基材涂覆杂化膜液的电镜图(标尺为400μm),e4为以纸为基材涂覆杂化膜液的电镜图(标尺为100μm)。
图6为实施例7以玻璃为基材杂化膜表面元素分布图,其中,1为实施例7制得的以玻璃为基材杂化膜的xps曲线,2为实施例3乳液制得以玻璃为基材涂膜的xps曲线,3为对比施例1乳液制得以玻璃为基材涂膜的xps曲线。
图7为实施例7制得以棉布为基材杂化膜防污图。
图8为实施例7以棉布为基材杂化膜防腐图,其中,a为不同ph值水溶液与杂化膜的接触角的曲线图,b为杂化膜对ph值分别为1、7和14时水溶液防水效果图。
图9为实施例7尼龙网为基材的杂化膜的油水分离效果图。
图10为实施例7尼龙网为基材的杂化膜的油水分离效率图。
具体实施方式
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
实施例12-(全氟1,1-二甲基-丁基)乙基丙烯酸酯制备
化合物2的合成及表征
300ml封管中依次加入kf(7.08g,0.12mol,1.2equiv.)、化合物1(30.00g,0.10mol,1.0equiv.)、溴乙酸乙酯(18.37g,0.11mol,1.1equiv.)、四丁基溴化铵(0.50g,1.5mmol,0.015equiv.)和100ml无水dmf,密封后置于80℃油浴锅中搅拌反应24h。冷却至室温后将反应体系倒入乙醚中,乙醚液依次用去离子水和饱和食盐水洗涤,无水硫酸钠干燥,过滤,浓缩,柱层析(etoac:petroleum=1:20)分离提纯,得无色液体产物31.6g,产率为78%。lcms(ei)m/z(%):361.1(100),406.1(2.1).
1hnmr(300mhz,cdcl3)δ4.30–4.06(m,2h),3.12(s,2h),1.24(t,j=7.1hz,3h).
19fnmr(282mhz,cdcl3)δ-63.24(p,j=10.8hz,6f),-80.64(t,j=13.3hz,3f),-106.99–-108.13(m,2f),-123.09(tt,j=21.2,10.5hz,2f)。
化合物3的合成及表征
250ml三口烧瓶中加入2.4glialh4(0.63mol),氩气置换三次后加入40ml无水乙醚,将21.4g化合物1(0.053mol)溶于60ml无水乙醚中,0℃下通过恒压滴液漏斗逐滴加入反应瓶中。室温下搅拌3h,升温至40℃反应4h。冷却至室温后,十水硫酸钠淬灭反应,硅藻土过滤,滤液依次用去离子水和饱和食盐水洗涤,无水硫酸钠干燥,过滤。减压蒸馏,收集70-73℃(35mmhg)馏分,得到12.9g无色液体,产率为67%。
lcms(ei)m/z(%):31.1(100),363.1(2.5)。
1hnmr(300mhz,acetone-d6)δ4.39–4.16(m,1h),3.86(dd,j=13.4,6.7hz,2h),2.51(t,j=7.6hz,2h)。
19fnmr(282mhz,acetone-d6)δ-65.24(p,j=11.4hz,6f),-81.82(t,j=13.8hz,3f),-108.79(tt,j=24.6,12.3hz,2f),-123.88–-124.77(m,2f)。
化合物4的合成及表征
50ml三口瓶,氩气保护下,依次加入10.0ml二氯甲烷、3.64g化合物2(0.010mol,1.0equiv.)和1.66ml三乙胺(0.012mol,1.2equiv.),降温至0℃,搅拌下缓慢加入0.87ml丙烯酰氯(0.011mol,1.1equiv.),室温反应8h。冰水浴下,加入5%硫酸水溶液淬灭反应,饱和氯化铵洗涤有机相,无水硫酸钠干燥,过滤,旋干,浓缩,柱层析(etoac:petroleum=1:20)分离提纯,得无色液体产物2-(全氟1,1-二甲基-丁基)乙基丙烯酸酯3.46g,产率为83%。
lcms(ei)m/z(%):55(100),249(7.74),418(3.94)。
1hnmr(400mhz,cdcl3)δ6.44(d,j=17.3hz,1h),6.11(dd,j=17.3,10.5hz,1h),5.88(d,j=10.4hz,1h),4.43(t,j=7.7hz,2h),2.57(t,j=7.7hz,2h)。
19fnmr(376mhz,cdcl3)δ-64.05(p,j=11.3hz,6f),-80.24(t,j=13.6hz,3f),-107.86(tt,j=24.4,12.2hz,2f),-123.01–-123.82(m,2f)。
实施例2
(1)在10ml反应瓶中依次加入去离子水1.5g,甲基丙烯酸甲酯0.328g,甲基丙烯酸丁酯1.232g,乳化剂dns-860.04g,室温下剧烈搅拌0.5小时形成预乳化液ⅰ;在10ml反应瓶中依次加入去离子水1.5g,甲基丙烯酸甲酯0.396g,甲基丙烯酸丁酯0.528g,丙烯酸十八酯0.5g,丙烯酸羟乙酯0.035g,2-(全氟1,1-二甲基-丁基)乙基丙烯酸酯0.2g,乳化剂dns-860.04g,室温下剧烈搅拌0.5小时形成预乳液ⅱ。在10ml反应瓶中依次加入去离子水2.7g与过硫酸铵0.04g,室温下溶解。
(2)20ml三口瓶中加入0.04gdns-86,氮气保护下加入1/3引发剂水溶液、1/3预乳化液ⅰ和1.5ml蒸馏水,在50℃温度下以250rpm转速搅拌20分钟,体系引发成功,出现蓝相。逐渐升温至70℃,逐滴缓慢加入剩余的2/3预乳化液ⅰ和1/3引发剂水溶液,滴加时间为90分钟。滴加完毕后,体系继续搅拌反应2小时。将预乳化液ⅱ与剩余的1/3aps水溶液缓慢滴加入反应体系中,滴加时间控制在2小时。滴加完毕后,体系继续搅拌反应2小时。随后,降温至40℃,加入适量氨水调节体系ph至7-9。收率92%,固含量为27.3%。
实施例3
(1)在10ml反应瓶中依次加入去离子水1.5g,甲基丙烯酸甲酯0.328g,甲基丙烯酸丁酯1.232g,乳化剂dns-860.04g,室温下剧烈搅拌0.5小时形成预乳化液ⅰ;在10ml反应瓶中依次加入去离子水1.5g,甲基丙烯酸甲酯0.396g,甲基丙烯酸丁酯0.528g,丙烯酸十八酯0.5g,丙烯酸羟乙酯0.035g,2-(全氟1,1-二甲基-丁基)乙基丙烯酸酯0.4g,乳化剂dns-860.04g,室温下剧烈搅拌0.5小时形成预乳液ⅱ。在10ml反应瓶中依次加入去离子水4g与过硫酸铵0.04g,室温下溶解。
(2)20ml三口瓶中加入0.04gdns-86,氮气保护下加入1/3引发剂水溶液、1/3预乳化液ⅰ和1.5ml蒸馏水,在60℃温度下以250rpm转速搅拌20分钟,体系引发成功,出现蓝相。逐渐升温至80℃,逐滴缓慢加入剩余的2/3预乳化液ⅰ和1/3引发剂水溶液,滴加时间为90分钟。滴加完毕后,体系继续搅拌反应6小时。将预乳化液ⅱ与剩余的1/3aps水溶液缓慢滴加入反应体系中,滴加时间控制在2小时。滴加完毕后,体系继续搅拌反应6小时。随后,降温至40℃,加入适量氨水调节体系ph至7-9。收率90%,固含量为31.1%。
实施例4
(1)在10ml反应瓶中依次加入去离子水1.5g,甲基丙烯酸甲酯0.328g,甲基丙烯酸丁酯1.232g,乳化剂dns-860.04g,室温下剧烈搅拌0.5小时形成预乳化液ⅰ;在10ml反应瓶中依次加入去离子水1.5g,甲基丙烯酸甲酯0.396g,甲基丙烯酸丁酯0.528g,丙烯酸十八酯0.5g,丙烯酸羟乙酯0.035g,2-(全氟1,1-二甲基-丁基)乙基丙烯酸酯0.6g,乳化剂dns-860.04g,室温下剧烈搅拌0.5小时形成预乳液ⅱ。在10ml反应瓶中依次加入去离子水4g与过硫酸铵0.04g,室温下溶解。
(2)20ml三口瓶中加入0.04gdns-86,氮气保护下加入1/3引发剂水溶液、1/3预乳化液ⅰ和1.5ml蒸馏水,在60℃温度下以250rpm转速搅拌20分钟,体系引发成功,出现蓝相。逐渐升温至80℃,逐滴缓慢加入剩余的2/3预乳化液ⅰ和1/3引发剂水溶液,滴加时间为90分钟。滴加完毕后,体系继续搅拌反应6小时。将预乳化液ⅱ与剩余的1/3aps水溶液缓慢滴加入反应体系中,滴加时间控制在2小时。滴加完毕后,体系继续搅拌反应6小时。随后,降温至40℃,加入适量氨水调节体系ph至7-9。收率93%,固含量为30.5%。
实施例5
(1)在10ml反应瓶中依次加入去离子水1.5g,甲基丙烯酸甲酯0.328g,甲基丙烯酸丁酯1.232g,乳化剂dns-860.04g,室温下剧烈搅拌0.5小时形成预乳化液ⅰ;在10ml反应瓶中依次加入去离子水1.5g,甲基丙烯酸甲酯0.396g,甲基丙烯酸丁酯0.528g,丙烯酸十八酯0.5g,丙烯酸羟乙酯0.035g,2-(全氟1,1-二甲基-丁基)乙基丙烯酸酯0.8g,乳化剂dns-860.04g,室温下剧烈搅拌0.5小时形成预乳液ⅱ。在10ml反应瓶中依次加入去离子水4g与过硫酸铵0.04g,室温下溶解。
(2)20ml三口瓶中加入0.04gdns-86,氮气保护下加入1/3引发剂水溶液、1/3预乳化液ⅰ和1.5ml蒸馏水,在60℃温度下以250rpm转速搅拌20分钟,体系引发成功,出现蓝相。逐渐升温至80℃,逐滴缓慢加入剩余的2/3预乳化液ⅰ和1/3引发剂水溶液,滴加时间为90分钟。滴加完毕后,体系继续搅拌反应6小时。将预乳化液ⅱ与剩余的1/3aps水溶液缓慢滴加入反应体系中,滴加时间控制在2小时。滴加完毕后,体系继续搅拌反应6小时。随后,降温至40℃,加入适量氨水调节体系ph至7-9。收率89%,固含量为32.8%。
实施例6
将粒径为15±5nm无机亲水二氧化硅配制成浓度为质量百分比为10%水溶液。5ml在容器中依次加入实施例2所得的乳液0.3g、去离子水0.7g、质量百分比为12%水性异氰酸酯0.03g,质量百分比为10%无机亲水二氧化硅水溶液0.4g,超声10-30min混合均匀。将混合后的乳液置于喷笔中,喷笔口径2mm,喷笔口与棉布基底材料水平距离为20cm,压力为200kpa。喷涂完毕后,基底材料置于80-100℃烘箱中固化1小时,得到棉布基材杂化膜材料。
实施例7
将粒径为15±5nm无机亲水二氧化硅配制成浓度为质量百分比为10%水溶液。5ml在容器中依次加入实施3所得乳液0.3g、去离子水0.7g、质量百分比为12%水性固化剂0.03g、质量百分比为10%无机亲水二氧化硅水溶液0.4g,超声10-30min混合均匀。将混合后的乳液置于喷笔中,喷笔口径2mm,喷笔口与棉布、钢网(400目)、尼龙网(400目)和纸张为基底材料水平距离为20cm,压力为200kpa。喷涂完毕后,基底材料置于80-100℃烘箱中固化1小时,得到以棉布、钢网(400目)、尼龙网(400目)和纸张为基材的杂化膜材料。
实施例8
将粒径为20nm无机亲水二氧化钛配制成浓度为质量百分比为10%水溶液。5ml在容器中依次加入实施例4所得乳液0.3g、去离子水0.7g、质量百分比为12%水性固化剂0.03g、质量百分比为10%无机亲水二氧化钛水溶液0.4g,超声10-30min混合均匀。将混合后的乳液置于喷笔中,喷笔口径2mm,喷笔口与基底材料水平距离为30cm,压力为200kpa。喷涂完毕后,基底材料置于80-100℃烘箱中固化1小时,得到杂化膜材料。
实施例9
将实施例5所得乳液参照实施例7制备杂化膜材料。
实施例10含氟聚丙烯酸酯乳液乳胶粒粒径表征
样品:实施例3中所得的含氟聚丙烯酸酯乳液
测试方法:将乳液稀释十倍后,取一定量稀释样品于激光粒度仪中进行测试。
图1中,1为核乳液粒子分布曲线,2为壳乳液粒子分布曲线。由图1可知,核乳液粒子与核-壳乳液粒子均呈球形均匀分布于乳液中,核乳液粒子的平均粒径为90.5nm。在核乳液加入壳层预乳液后,壳乳液中含氟单体与其他单体成功聚合到核乳液中的粒子上,形成核-壳结构,壳层粒径增大至102.2nm。
实施例11含氟聚丙烯酸酯乳液乳核-壳结构表征
样品:实施例3中所得的含氟聚丙烯酸酯乳液
测试方法:将乳液稀释十倍后,取一定量稀释样品于铜网中进行透射电镜测试。
图2中,(a)为未经染色核乳液,(b)经染色核乳液,(c)未经染色核-壳乳液,(d)经染色核-壳乳液。由图2可知,从透射电镜照片可看出粒径大小大致在100nm上下,与激光粒度仪所测结果一致。此外,由于电子在含氟壳相与非含氟核相的透过性不同,核-壳乳液样品经质量百分比为1%磷钨酸染色后,可以明显的观察到核-壳结构,如图2(d)所示,其中,较暗的区域为核相,较亮的区域为含氟的壳相。证实本发明制得核壳型共聚物乳液具有核-壳结构,其中壳层为含氟的聚合物乳液。
实施例12
将实施例6制得的以棉布为基材的杂化膜,该杂化膜由实施例2乳液制得,实施例2制得乳液,含氟丙烯酸酯单体占总单体质量的质量百分为6.2%。
将实施例6制得杂化膜与水接触,杂化膜与水的接触角由接触角测量仪测定,其经涂膜后与水的接触角为大于150°,表现为超疏水性,如图3所示,经整理后的棉布具有了拒水的性能。
对比参考文献1(cn105440201a)公开了一种具有核壳结构的共聚乳液;对比参考文献2(张庆华,詹晓力,陈丰秋.氟代丙烯酸醋三元共聚物细乳液的合成与表征[j].高等学校化学学报,2005,26(3):575-579.)公开了一种无规共聚乳液。上述两篇参考文献中,考察了含氟丙烯酸酯单体用量对乳胶膜拒水拒油性能的影响,具体地如表1所示。
当以接触角做为评价拒水拒油的指标时,由表1可知,本申请的核壳结构共聚乳液在满足生物降解的同时,即使用含氟丙烯酸酯单体中碳氟原子数为6个,所用的含氟丙烯酸酯单体与总单体质量的质量百分比为6.2%,低于对比参考文献1(cn105440201a)中8.5%;由其制得的涂膜与水接触角大于150°,高于对比参考文献1(cn105440201a)中与水接触角124.6°,表现了超疏水性,其拒水效果优于现有技术的拒水效果。
表1本申请核壳型共聚物的乳液与现有技术共聚物乳液对比
实施例13
将实施例7所得的棉布、钢网(400目)、尼龙网(400目)和纸张为基底材料的杂化膜分别与水接触,涂膜与水的接触角由接触角测量仪测定,其接触角如图4所示,图4a为基底材料为棉布的杂化膜与水的接触角,图4b为基底材料为钢网(400目)的杂化膜与水的接触角,图4c为基底材料为尼龙网(400目)的杂化膜与水的接触角,图4d为基底材料为纸张的杂化膜与水的接触角,四种基底材料经涂膜后与水的接触角均大于150°,均具有了超疏水的性能。可见,四种材料经涂膜后均具有拒水性性能,聚合物乳液在制备杂化膜时具有广泛的底物适用性。
实施例14
扫描电镜分别对玻璃、棉布、钢网、尼龙网、纸张基材以及实施例7所得杂化膜后基底材料进行扫描。图5中:
a1为以玻璃为基材涂覆核-壳聚合物乳液的电镜图(标尺为50μm),a2为以玻璃为基材涂覆核-壳聚合物乳液的电镜图(标尺为10μm),a3为以玻璃为基材涂覆杂化膜液的电镜图(标尺为50μm),a4为以玻璃为基材涂覆杂化膜液的电镜图(标尺为10μm);
b1为以棉布为基材的电镜图(标尺为400μm),b2为以棉布为基材的电镜图(标尺为100μm),b3为以棉布为基材涂覆杂化膜液的电镜图(标尺为400μm),b4为以棉布为基材涂覆杂化膜液的电镜图(标尺为100μm);
c1为钢网为基材的电镜图(标尺为400μm),c2为钢网基材的电镜图(标尺为100μm),c3为以钢网为基材涂覆杂化膜液的电镜图(标尺为400μm),c4为以钢网为基材涂覆杂化膜液的电镜图(标尺为100μm);
d1为尼龙网基材的电镜图(标尺为400μm),d2为尼龙网基材的电镜图(标尺为100μm),d3为以尼龙网为基材涂覆杂化膜液的电镜图(标尺为400μm),d4为以尼龙网为基材涂覆杂化膜液的电镜图(标尺为100μm);
e1为纸为基材的电镜图(标尺为400μm),e2为纸为基材的电镜图(标尺为100μm),e3为以纸为基材涂覆杂化膜液的电镜图(标尺为400μm),e4为以纸为基材涂覆杂化膜液的电镜图(标尺为100μm)。
从图5可以看出,在未经涂膜前,基底材料表面十分光滑,经过涂膜后,材料表面具有类似山丘一样的突起结构。从图a3和a4中可以清晰的看到经涂膜覆盖后的玻璃表面具有微纳米多尺度的突起表面,该突起表面可赋予其超疏水的性能。另外,从图b、c和d可知,含微孔的软性材料经涂膜后,涂膜材料不会堵住材料本身的微孔,即不会改变材料本身具有的某些性能,例如透气性。
实施例15
将实施例7制得的以玻璃为基材杂化膜,实施例3和对比实施例1制得乳液各自独立地在玻璃基材上涂膜。对其涂膜表面的元素分析,其具体的元素含量如表2所示。从表2可以看出,经实施例7制得的以玻璃为基材杂化膜表面的氟元素实测值为20.6%,而实施例3制得乳液中氟元素的其理论值应为5.6%,说明在成膜过程中,聚合物中的含氟片段更倾向于向表面聚集,从而降低膜表面的表面自由能,达到更好的疏水效果。实施例7制得的以玻璃为基材杂化膜中含有0.12%的纳米二氧化硅粒子后,膜表面氟元素的含量降低至0.8%,硅元素的含量为12.4%,此现象主要是由于x射线光电子能谱(xps)的探测深度通常为2-10nm,而所加入的纳米二氧化硅的粒径为15±5nm,该现象说明纳米二氧化硅在膜表面的分布十分均匀,因此难以探测得氟元素的信号。
表2涂膜表面元素组成
图6中,1为实施例7制得的以玻璃为基材杂化膜的xps曲线,2为实施例3乳液制得以玻璃为基材涂膜的xps曲线,3为对比施例1乳液制得以玻璃为基材涂膜的xps曲线。如图6所示,从涂膜的xps曲线上可知,在688、529、283和100ev分别出现f1s、o1s、c1s和si2p的特征信号峰;从图6中还可知,由对比对比实施例1制得乳液制备的涂膜的xps曲线上可以看到硅元素的si2p特征信号,该信号应为基底材料载玻片上的硅原子所致,此现象主要是由于退湿现象导致的涂膜不均匀。在实施例7制得的以玻璃为基材杂化膜的xps曲线中并未观察到该现象,表明本发明的含氟共聚物乳液可提高成膜均匀性。
实施例16
将不同的液体水、果汁、牛奶、酒、墨水和咖啡滴在实施例7所得的以棉布为基材杂化膜上,从图7可以看出,不同液体在其表面均聚集为球状的液滴,无法渗透入棉布内部,说明经核壳型共聚物的乳液整理后的棉布具有了拒污性能。
实施例17
将具有不同ph值得水溶液与实施例7所得的以棉布为基材杂化膜的接触,从图8a中可知,经整理后棉布分别与酸性溶液和碱性溶液接触,其接触角均大于150°,在ph值分别为1、7、14时,水溶液均呈现出液滴形态,如图8b所示。由此表明,经核壳型共聚物的乳液整理后的棉布具有拒水性能,同时还具有防酸和防碱的防腐性能。
实施例18
将水和正十六烷分别滴加在实施例7所得的涂有杂化膜的尼龙网,尼龙网状材料表现为超疏水状态,而十六烷却可以瞬间从尼龙网上渗透下去。利用杂化膜在尼龙网的这个特殊性质,将水和二氯甲烷混合溶液同时倒入配有涂有杂化膜的尼龙网的分离装置中,利用重力驱动,探究尼龙网的油水分离功能。结果如图9所示,为了方便区分,将油用苏丹ii染成红色,水用亚甲基蓝染成蓝色。由图9可以看出,二氯甲烷在重力驱动下,迅速透过尼龙网,而水始终在尼龙网上方,说明经涂膜后的尼龙网具有油水分离的功能。如图10所示,通过称重法测得分离效率98%上下,且重复20次未见效率降低。目前,本申请是首例利用含氟丙烯酸酯聚合物杂化膜实现了油水分离的功能。
对比实施例1
(1)在10ml反应瓶中依次加入去离子水1.5g,甲基丙烯酸甲酯0.328g,甲基丙烯酸丁酯1.232g,乳化剂dns-860.04g,室温下剧烈搅拌0.5小时形成预乳化液ⅰ;在10ml反应瓶中依次加入去离子水1.5g,甲基丙烯酸甲酯0.396g,甲基丙烯酸丁酯0.528g,丙烯酸十八酯0.5g,丙烯酸羟乙酯0.035g,乳化剂dns-860.04g,室温下剧烈搅拌0.5小时形成预乳液ⅱ。在10ml反应瓶中依次加入去离子水4g与过硫酸铵0.04g,室温下溶解。
(2)20ml三口瓶中加入0.04gdns-86,氮气保护下加入1/3引发剂水溶液、1/3预乳化液ⅰ和1.5ml蒸馏水,在60℃温度下以250rpm转速搅拌20分钟,体系引发成功,出现蓝相。逐渐升温至80℃,逐滴缓慢加入剩余的2/3预乳化液ⅰ和1/3引发剂水溶液,滴加时间为90分钟。滴加完毕后,体系继续搅拌反应6小时。将预乳化液ⅱ与剩余的1/3aps水溶液缓慢滴加入反应体系中,滴加时间控制在2小时。滴加完毕后,体系继续搅拌反应6小时。随后,降温至40℃,加入适量氨水调节体系ph至7-9。收率97%,固含量为30.8%。
- 还没有人留言评论。精彩留言会获得点赞!