一种高降解DNA测序文库的构建方法及其应用与流程
本发明属于生物医学技术领域,具体涉及一种高降解dna测序文库的构建方法及其应用。
背景技术
自下一代测序(next-generationsequencing,ngs)技术诞生以来,该技术成为一种重要的研究及检测、诊断技术,对生命科学、生物医学及医学的研究产生了深刻地影响。随着计算机运算能力的发展,ngs平台推动了过去几年生物学知识的爆炸式增长。与传统的sanger测序法相比,ngs在测序前需要制备测序文库。ngs的测序过程已经高度自动化,而测序文库的制备是ngs测序的关键。
现有的标准建库流程主要步骤包括:dna片段化(超声或酶切)、末端修平、加a、y接头连接、片段选择及pcr扩增。标准建库流程为了提高接头连接步骤的效率,须对dna片段进行多酶混合物处理的末端修平,使dna片段的两端均成为平末端,以及必不可少的末端加a处理。虽然有不少公司推出了这些步骤的合并或优化方法,也推出了类似y接头(如颈环接头)的接头连接等方法,但建库技术没有从根本上得到改变。此外,该种建库方法中,广泛采用了y型接头,直到最后的pcr步骤才用带有index的pcr引物进行不同dna样品的区分,之后混合进行同一通道(lane)的测序。这种每个dna样品单独经历整个建库建库流程才能混合测序的方法,极大地增大了操作的复杂性、试剂及人力消耗,不仅建库成本高,而且易于造成不同dna样品在建库期间的人为偏差(bias),不利于不同样品间的测序结果的平行比较。基于y型接头的标准建库流程以及近年来出现的基于tn5酶的片段化接头连接技术(tagmentation),都比较适合分子量大的dna的建库测序,如从各种细胞中提取的高分子量基因组dna等。
高降解dna常常是自然发生的降解程度较高的dna,如常见的血液游离dna、循环肿瘤dna、循环胎儿dna、古生物dna、法医dna、水体等环境游离dna等。这类dna由于脱离自然的细胞内环境,受到各种理化因素的作用而发生断裂,成为分子量小且分子内磷酸二酯键受损断裂的带有切刻(nick)的dna分子,因此样品内分子具有多样性,包含了由于高度降解而在两条链中产生裂痕的超短(<100bp)双链dna(dsdna)、单链dna(ssdna)和常规dsdna。运用y型接头标准建库流程及tn5片段化接头连接技术对这类dna建库时,难以建立样品保真性高的dna文库,导致带来很多dna序列信息丢失。
为了解决这一问题,近年来逐渐发展了基于ssdna的ngs文库构建方法。基于ssdna的ngs文库构建方法在建库时,都首先将dna样品变性解链,使其成为由各种长度ssdna构成的dna样品。ssdna文库制备方法构建的文库中包含低于100bp的dna片段,能够使原样品中的各种dna分子有相同的机会进入文库被测序,因此提供更丰富的信息。然而,目前的基于ssdna的方法通常涉及多个耗时且高成本低效率的步骤,包括起始dna去磷酸化,通过生物素磁珠分离纯化,或通过特殊的酶(如circligaseii)进行单链接头的连接。即使使用商品化的试剂盒,也需要特殊的逆转录病毒逆转录酶(dnasmartchip-seqkit,clontech)。因此,基于ssdna的ngs文库构建方法仍需发展新的技术。
血液游离dna(cfdna)是一种典型的高降解dna。cfdna主要来源于核基因组,其片段长度集中在166bp左右,为围绕一个组蛋白八聚体缠绕的dna片段的长度。然而,cfdna具有丰富的多样性,包括由于高度降解而在两条链中产生裂痕的超短(<100bp)dsdna,单链dna(ssdna)和常规dsdna。因此,基于ssdna的ngs文库构建方法非常适合cfdna的建库测序。cfdna的ngs建库测序具有重要的临床研究和诊断价值,成为目前无创检测(nid)、液体活检(liquidbiopsy)、产前诊断(nipt)、体外诊断(ivd)、床旁诊断(poct)等领域的主要研究对象。
技术实现要素:
解决的技术问题:针对上述现有基于ssdna的ngs文库构建方法的缺点,本发明提供了一种基于单链接头文库制备技术salp的高降解dna测序文库的构建新方法及其应用,该方法极大的简化了基于ssdna的ngs文库制备的过程,实现了低成本、高效率、高通量和低偏倚的高降解dna测序文库构建。
技术方案:一种高降解dna测序文库的构建方法,包括以下步骤:
步骤1,将双链dna1(dsdna)变性,使其成为单链dna1(ssdna);
步骤2,在步骤1所得单链dna1的3′端连接单链接头(singlestrandadaptor,ssa),成为单链dna2;
步骤3,用dna聚合酶延伸步骤2所得单链dna2使其成为双链dna2;
步骤4,向步骤3所得双链dna2无单链接头的一端连接标签t接头(barcodetadaptor,bta),成为双链dna3;
步骤5,pcr扩增步骤4两端连接接头的双链dna3使其成为可测序文库。
进一步地,步骤1中所述双链dna1为因脱离自然细胞内环境而受到各种非人为理化因素作用而发生高度断裂的dna片段或人为降解得到的dsdna片段。
因脱离自然细胞内环境而受到各种非人为理化因素作用而发生高度断裂的dna片段,可以是血液游离dna(cfdna)、循环肿瘤dna(ctdna)、循环胎儿dna(cffdna)、古生物dna、法医dna或水体等环境游离dna。
人为降解得到的dsdna片段,可以是超声波剪切的dna片段、酶切产生的dna片段或基于转座体片段化产生的dna片段。
进一步地,步骤2中所述单链接头为带有粘性末端的双链寡核苷酸1,该双链寡核苷酸1由寡核苷酸1和寡核苷酸2退火形成;其中寡核苷酸1从5′端到3′端的序列结构为:5′端羟基→恒定序列→随机核苷酸序列→3′端羟基,寡苷酸2从5′端到3′端的序列结构为:5′端磷酸基团→恒定序列→3′端氨基,且寡苷酸2的恒定序列与寡核苷酸1的恒定序列碱基长度相同且全序列反向互补。
进一步地,所述随机核苷酸序列为包括1-4个碱基的核苷酸序列。作为优选,随机核苷酸序列为3个碱基的核苷酸序列,3个碱基选自a、t、c或g。
进一步地,寡核苷酸1的优选序列为:5′-acactctttccctacacgacgctcttccgatctnnn-3′(表2)。
进一步地,寡核苷酸2的优选序列为:5′-[phos]-agatcggaagagcgtcgtgtagggaaagagtgt-[nh2]-3′(表2);其中[phos]为磷酸基团,[nh2]为氨基基团。
进一步地,所述粘性末端可与单链dna的3′端退火,并可通过核酸连接酶催化5′端磷酸基团与单链dna3′端羟基基团形成3′-5′磷酸二酯键;所述核酸连接酶为t4dna连接酶。
所述dna聚合酶可以是各种dna聚合酶。若所述dna聚合酶为普通taqdna聚合酶,则步骤3产生的双链dna的3′端末端自然产生一个突出的a碱基,则dna聚合酶延伸产物可直接用于步骤4连接t接头;所述dna聚合酶若为其他高保真dna聚合酶,由于步骤3产生的双链dna的3′端末端不出现一个突出的a碱基,则延伸产物需再用普通taqdna聚合酶及其他具有类似功能的酶处理,使延伸产物的3′端末端产生一个突出的a碱基,再用于步骤4连接t接头。
进一步地,所述标签t接头为带有粘性末端的双链寡核苷酸2,该双链寡核苷酸2由寡核苷酸3和寡核苷酸4退火形成;其中寡核苷酸3从5′端到3′端的序列结构为:5′端羟基→恒定序列1→标签序列→恒定序列2→一个t碱基→3′端羟基,寡苷酸4从5′端到3′端的序列结构为:5′端磷酸基团→恒定序列→3′端羟基,且寡苷酸4的恒定序列与寡核苷酸3的恒定序列2碱基长度相同且全序列反向互补。
进一步地,寡核苷酸3的恒定序列1的序列为:5′-gactggagttcagacgtgtgctcttccgatct-3′(表3);恒定序列2的优选序列为:5′-agatgtgtataagagacagt-3′(表3);标签序列为6个碱基的可变序列(表3)。
进一步地,寡核苷酸4的恒定序列的优选序列为:5′-ctgtctcttatacacatct-3′(表3)。
进一步地,所述标签t接头,其一个粘性末端为3′端突出一个t碱基,该t碱基可与步骤3产生的双链dna的3′端突出a碱基退火;再由核酸连接酶催化t接头与步骤3产生的双链dna间形成3′-5′磷酸二酯键,所述核酸连接酶一般为t4dna连接酶。
所述标签t接头为从成本最低角度设计的最佳结构及末端修饰,其中寡核苷酸3的5′端和3′端均为寡核酸固相化学合成时自然产生的羟基,无需额外修饰,成本最低;其中寡核苷酸4的5′端为修饰的磷酸基团,而3′端为寡核酸固相化学合成时自然产生的羟基;寡核苷酸4的5′端磷酸基团参与标签t接头与步骤3产生的双链dna连接时形成3′-5′磷酸二酯键。
进一步地,步骤5中pcr扩增是以单链接头和标签t接头为退火位点。
进一步地,pcr扩增的引物序列分别为:5′-aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatct-3′(表4)、5′-caagcagaagacggcatacgagattctgacatgtgactggagttcagacgtgtgctcttccgatct-3′(表4);其中tgacat为索引序列(index),此引物可用illumina其他index引物替代。
为了发展优越性更好的基于ssdna的测序文库构建方法,本发明以cfdna为研究对象,提供了一种基于单链接头文库制备方法salp技术的高降解dna测序文库构建新方法。基于单链接头文库制备技术salp的高降解dna测序文库构建新方法无需对样品dna进行去磷酸化、末端修复、修饰等预处理,也无需使用磁珠捕获,极大的简化了基于ssdna的ngs文库制备的过程。本发明提出的高降解dna测序文库构建新方法,仅需使用少数几个用于制备接头的化学修饰的寡核苷酸,以及两种常见酶,t4dna连接酶和taq聚合酶,极大降低了实验成本。同时,本发明新方法通过使用已有的单链接头(ssa)和带有标签(barcode)的特殊t接头(bta),实现了低成本、高效率、高通量和低偏倚的高降解dna测序文库构建。
所述高降解dna测序文库构建方法,该程序中,若要同时对多个dna样品进行建库测序,可采用该程序的高通量建库流程,其实验流程为:①将各样品dsdna片段变性,使其成为ssdna;②在各样品ssdna的3′端分别连接一种通用ssa;③对连接单链接头的ssdna用dna聚合酶延伸,使其成为dsdna;④不同样品连接带有不同标签序列的bta;⑤将连接了bta的各样品dsdna混合,形成一混合dna样品;⑥pcr扩增两端连接接头(ssa及bta)的dsdna,使其成为ngs可测序的dna文库。该程序中,将带有不同bta的不同dna样品混合,成为一个dna混合物,作为一个dna混合样品,进行pcr扩增,可简化了多dna样品的建库操作、消除了pcr扩增步骤可能带来的偏差(bias),便于不同dna样品间序列信息的比较分析。
本发明所述的高降解dna测序文库的构建方法在无创检测(nid)、液体活检(liquidbiopsy)、产前诊断(nipt)、体外诊断(ivd)、床旁诊断(poct)、法医鉴定、环境生物检测、环境生物调查、考古等领域的主要研究对象。
有益效果:本发明提供了一种高降解dna测序文库的构建方法,该方法具有几个显著优点:(1)该方法可对任何高降解dna片段不经任何修饰处理,即可进行建库。(2)该方法设计并使用特殊的单链接头(ssa)和标签t接头(bta),用于从单链dna开始进行dna测序文库的构建,避免双链建库法遗漏dna样品特别是高降解dna样品的部分信息。(3)标签t接头(bta)的使用可使多样品dna建库混合后同步pcr扩增,有利于简化了建库操作、降低试剂及人工消耗、避免了建库中pcr扩增在不同样品间可能产生的偏差,非常有利于多样本快速建库及不同样本之间测序信息的比较分析。(4)本发明提出的高降解dna测序文库的构建方法,其接头序列的设计是技术关键,所用接头序列、结构及修饰为制备成本最低、通用性最佳的设计。(5)本发明提出的高降解dna测序文库的构建方法只需t4dna连接酶、taqdna聚合酶等常用低价酶材料,避免了目前单链建库存在的弊病(如特殊酶的需要、接头连接效率低等)。
附图说明
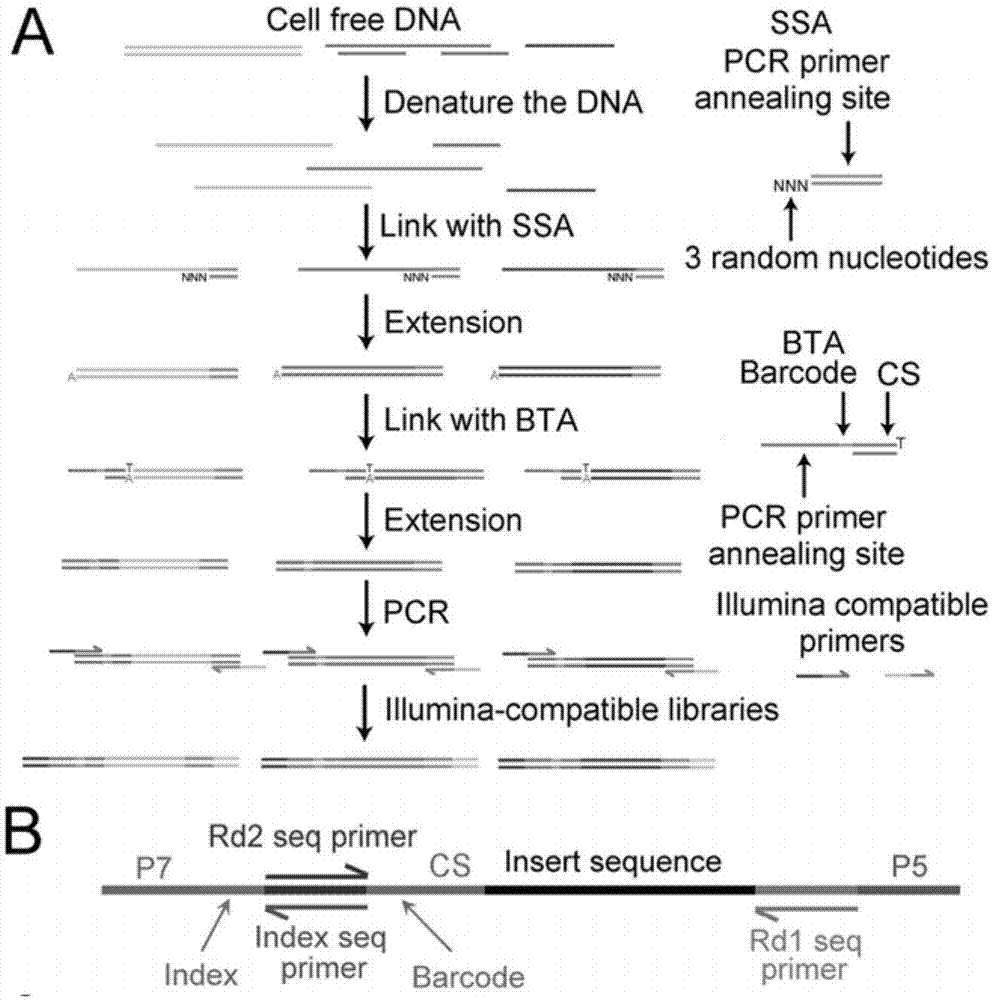
图1为基于salp技术的cfdna文库构建原理及流程示意图。a为基于salp的cfdna文库构建流程,单链接头(ssa)是在其3'末端具有3个随机核苷酸(3n)突出端的双链寡核苷酸,barcodet接头(bta)含有3't突出端,双链固定序列(cs)和由barcode和pcr引物退火位点组成的5'单链。b为通过改进的salp方法构建的cfdnaillumina测序兼容ngs文库结构。
图2为基于salp技术高通量制备多个cfdna样品的ngs文库流程。
图3为凝胶回收前后cfdna文库的琼脂糖凝胶电泳,l1和l2显示了两个cfdna文库。
图4为文库片段分布,由安捷伦2100高灵敏度dna芯片检测。
图5为通过salp鉴定出的不同样品中cfdna特征。a为reads密度的分布,cfdna1~20的reads密度由外向内依次显示,reads密度指1-mb基因组窗口中的reads数量,箭头2表示reads密度分布相同的基因组区域,箭头1表示reads密度存在差异的基因组区域。b为三种类型cfdna的平均reads密度,c为三种cfdna的平均reads长度,d为三种cfdna的gc含量,其中,pre:术前患者cfdna样本;post:术后患者cfdna样本;normal:正常人cfdna样本。
图6为不同cfdna样品测序鉴定染色质开放状态。a为通过cfdna测序鉴定染色质开放状态原理。b为tss周围reads的分布,曲线图显示了tss周围区域的平均reads密度,计算并显示所有reads密度的log10值,其中,rc,reads数目;arc,平均reads数目。c为不同cfdna样品中具有显著差异的启动子区域reads密度热图。d为23个基因启动子区reads密度的热图。e为实施例1(23个基因)鉴定食管癌相关基因与文献报道比较。f为23个基因的go分析,图中显示了每个go条目的p值和包含的基因个数。g为23个食管癌相关基因的表达比较,箱线图显示了不同类型样品中基因的表达,热图显示染色质开放状态。g中箭头标示的基因是指文献报道的基因,未标记的基因是指新鉴定的基因,其他基因是指文献报道过的。
图7为特定基因启动子区域的快照(snapshot)。ucsc基因组浏览器显示5个基因的启动子区域。cfdnatrack、te7h3k27acchip-seqtrack、kyse510h3k27acchip-seqtrack分别展示。同时显示h3k27ac和dnacluster,以表明染色质区域的开放程度。te7与kyse510为两种食管癌细胞株。
图8为不同来源的cfdna的染色质状态比较。a为peak密度的分布,peak密度定义为基因组上每个1-mb窗口中的peak数。te7,kyse510和cfdna1至20的peak密度从外到内依次显示。b为食管癌患者cfdna和两种食管癌细胞系(te7和kyse510)h3k27acchip-seq重叠peak统计。c为不同类型cfdna位于启动子区的peak百分比。d为不同类型cfdnareads数目标准化后获得的peak数目,每个样品的reads数目标准化至106。e为不同样品中基因表达的分类标准。f为食管癌患者和健康人特异性表达基因比较。g为两类基因的go分析。
图9为cfdna测序鉴定的食管癌特异性表达基因与rna-seq鉴定的食管癌组织中差异表达基因比较。a为cfdna和rna-seq的差异基因比较。b为由cfdna和rna-seq鉴定的食管癌特异性表达基因富集得到的相同go条目。其中,bp:生物过程;mf:分子功能。从tcga下载来自184个食管癌和14个癌旁组织的rna-seq数据。用deseq2进行差异表达分析,将padj<0.05且表达倍数大于2的基因定义为差异表达基因。选择差异表达倍数最高的前1000个基因通过david进行go分析。
图10为不同cfdna样本中的突变。a为突变密度的分布,突变密度定义为基因组上每个1-mb窗口中的突变数,箭头2表示具有较高突变密度的基因组区域,箭头1表示具有较低突变密度的基因组区域,cfdna1至20的突变密度从外向内依次显示。b为不同cfdna样品中的msk-impactpanel基因比较,将用cfdna测序鉴定的具有外显子突变的基因与msk-impactpanel基因进行比较。c为与msk-impactpanel重叠的基因中的突变注释。d为28个术前样本特有基因go分析。
具体实施方式
以下结合附图和实施例对本发明作进一步说明。
实施例1血液游离dna建库测序
实验材料和方法
样本收集:在南京大学医学院附属金陵医院(中国南京)的帮助下共收集得到20份全血样本。其中,四份来源于健康人、十一份收集自术前食管癌患者,五份收集自术后食管癌患者(表1)。
表1.cfdna来源信息
游离dna提取:全血样品在4℃下以1,600g离心15分钟,将上清液转移至新的离心管。含有上清液的试管在4℃下以16,000g离心10分钟,上清液即为血浆。全部血浆储存-80℃备用。使用plasmacirculatingdna试剂盒(tiangen,dp339)以200μl血浆为起始物,用以分离cfdna,提取得到的cfdna溶解于20μltris-edta(te)缓冲液中,-20℃储存。
接头制备:所需寡核苷酸均由上海生工合成。制备单链接头(ssa),将ssa-pn-3n和ssa-pnrev(表2)溶解于ddh2o,终浓度为100μm,等摩尔混合于pcr管中。制备带有barcodet接头(bta),将带有不同barcode的寡核苷酸和bta-通用寡核苷酸(表3)分别溶解于ddh2o,终浓度为100μm,等摩尔混合于pcr管中。所有寡核苷酸混合物95℃的水浴变性5分钟后,逐渐冷却至25℃,退火形成相应接头。
表2.制备单链接头(ssa)的寡核苷酸
表3.制备标签t接头的寡核苷酸
用改进的salp方法制备cfdnangs文库:将7μlcfdna样品在95℃温育5分钟并立即在冰上孵育5分钟。变性的cfdna与ssa在10μl反应体系中16℃过夜连接,反应体系组分包括:1μlt4dna连接酶(neb,m0202l),1×t4dna连接酶缓冲液,0.5μmssa。将连接产物与10μl2×预混合taq聚合酶(takara,r004a)混合,72℃下孵育15分钟。以1.8×ampurexp磁珠(beckmancoulter)纯化产物。纯化产物与1μlt4dna连接酶,1×t4dna连接酶缓冲液,0.1μmbta混合,补加ddh2o至10μl,16℃连接2小时。1.8×ampurexp磁珠纯化后,连接产物于50μl体系中进行扩增,反应体系组分为1×
表4.文库制备pcr引物
ngs测序:用测序平台相容的引物扩增后共获得20个illumina测序平台兼容的文库(表4)。使用qubit2.0测定文库浓度,并以相同的dna质量(ng)混合,获得最终测序文库。通过agilentbioanalyzer2100高灵敏度dna芯片检测文库片段分布。使用illuminahiseqxten平台(南京世和)对文库进行测序。
cfdna测序数据分析:通过perl脚本根据barcode分割原始reads数据。从双端测序的read25'端去除固定序列(cs)(19bp)和barcode序列(6bp)。使用bowtie2将所有reads比对到人基因组(hg19)。为了确保长片段能够比对到基因组上,设定参数-x2000。用bcftools进行snv分析[30]。通过annovar使用默认参数对snv进行注释。利用david网站进行geneontology(go)分析。通过bedtools统计reads数目。deseq2分析不同样品中转录起始位点(transcriptionstartsite,tss)上游1kb区域的开放程度,选择其中p<0.05的区域为具有显著差异的区域。由geo数据库下载两种食管癌细胞系,te7和kyse510的h3k27acchip-seq数据,检索号为gse76861。通过bowtie2将chip-seq原始reads比对到hg19基因组。使用macs2进行peakcalling,homer进行peak的注释。所有track均由ucsc基因组浏览器展示。rna-seqfragmentsperkilobasespermillionreads(fkpm)数据由thecancergenomeatlas(tcga)数据库下载,包括163个食管癌和11个正常样品。使用perl脚本对选定基因的fkpm进行比较。
为了比较启动子区域中的snv,将tss上游10kb定义为基因的调控区域。筛选出术前样本中位于调控区的突变,以突变位置为中心上下游共截取20bp区域,通过fimo软件搜索该区域内被hocomoco(版本11)数据库收录的转录因子结合位点。
实验结果
1.运用改进的salp-seq构建cfdnangs文库
salp方法在构建降解dna的ngs文库时具有显着优势。为了使用salp方法构建cfdna的ngs文库,我们对其进行了改进以提高效率并降低成本。如图1a所示,特殊设计的barcodet接头(bta),包含由3't突出端,固定序列构成的双链区,以及由可变的6碱基barcode和固定的pcr引物退火位点构成的单链区。此设计有助于提高通量并简化过程。为了构建cfdna的ngs文库,将提取得到的cfdna变性为单链,然后与具有3个随机碱基突出端的ssa连接。用taq聚合酶延伸后,产生腺嘌呤(a)突出端,随后连接bta。在延伸步骤之后,通过可分别与ssa和bta退火的illumina兼容引物扩增文库(图1a)。文库的结构如图1b所示,可直接通过illumina平台进行测序。
为了验证改进的salp在构建cfdna文库中的效率,选取两个cfdna样品用改进的salp方法构建ngs文库。如图2所示,文库的片段大小主要集中于300bp左右,表明插入片段的长度约为180bp,与之前报道的cfdna长度一致。可以从琼脂糖凝胶中观察到阶梯状的条带,表明改进后的新方法能够灵敏地捕获不同长度的cfdna(图2)。
改进的salp-seq方法可用于制备含有多个cfdna样本的ngs文库,这对于有效分析大量临床血液样本非常有用。使用含有不同barcode的bta能够标记不同的样品(表2)。在bta连接步骤之前,所有cfdna样品分别单独进行处理(图1a),随后不同的cfdna样品通过bta连接步骤,与不同bta连接(图3)。bta连接后,可通过两种策略混合测序文库(图3)。一种是在bta连接后立即将标记后的样品进行混合,然后通过单管pcr扩增获得最终的illumina测序文库。另一种是针对不同样本单独扩增,然后合并扩增的文库并获得最终的illumina测序文库(图3)。使用第二种策略,我们构建了20个具有不同bta的cfdna文库。混合20个文库后,成功构建了illumina测序平台兼容的测序文库(图4)。最终的文库由illuminahiseqxten平台测序。获得了总共420,594,419条能够比对到参考基因组上的reads(表5)。
表5.测序结果reads统计
2.cfdna特征分析
作为液体活检的重要样本来源,cfdna具有十分重要的意义,可为诊断提供许多有用的线索。为了研究cfdna在全基因组水平上的分布,我们计算并标准化每个1-mb窗口中的reads密度。结果表明,不同样品中cfdna分布在整个基因组水平上有很大差异(图5a)。一些基因组区域在20个样本中具有相同水平的reads密度,而其他区域的reads密度则相对较大(图5a)。来自食管癌患者的cfdna具有最高的平均reads密度,但来自正常人的cfdna具有较低的平均reads密度(图5b)。这可能是由于基因和调控序列的拷贝异常增加所致。cfdna的长度与临床场景密切相关。为了比较不同样品的长度分布,我们计算了全基因组水平的cfdna长度。结果表明术前食管癌患者具有最短的cfdna,正常人具有较长的cfdna(图5c)。为了进一步表征不同类型样品中cfdna的特征,我们还计算了cfdna的gc含量(图5d)。结果显示术前食管癌患者的cfdna具有最低的gc含量,而来自正常人的cfdna具有最高的gc含量。手术提高了cfdna的gc含量。这些结果揭示了cfdna的reads密度,长度和gc含量这三个特征可以作为食管癌液体活检的诊断标记。
3.运用cfdna鉴定染色质状态
基于reads密度的全基因组分布(图5a),我们推断cfdna的ngs数据可以用来分析不同样本的染色质状态。只有核小体保护的基因组区域才能够进入cfdna的ngs测序环节(图6a)。为了验证该假设,以100bp为窗口,计算基因的tss上下游±5kb区域内的reads密度,并计算每个窗口的平均reads密度。结果显示在正常的cfdna统计结果中,tss附近出现一个峰,而在食管癌cfdna的统计结果中,tss附近有一个谷(图6b)。该结果表明cfdnangs能够用于检测染色质开放状态。为了进一步比较癌症和正常样品之间的染色质开放度,将tss上游1kb区域定义为启动子区(promoter),计算每个样品中所有启动子区的reads密度。结果显示,健康个体和食管癌患者cfdna的启动子reads密度之间存在很大差异(图6c)。值得注意的是,一些启动子在所有癌症样品中显示极低的reads密度,但在正常样品中显示较高密度(图6d)。共发现23个基因具有该特征。在文献研究之后,我们发现这些基因中有9个与食管癌密切相关(图6e)。因此我们推断剩余的14个基因是新鉴定的食管癌相关基因。典型基因启动子的ucsctrack也说明了癌症与正常样本之间的显着差异(图7),进一步验证了这一发现。这些基因的go分析显示,富集最显著的条目(chromosomeorganization)与染色体构象相关,表明这些基因中的含有在调节染色质结构中起关键作用的基因(图6f)。我们发现该go项由5个基因富集得到,包括ino80、whsc1、terf2ip、ncapd3和suv420h1,其中基因ino80和whsc1是已知的食管癌相关基因(表4)。其他富集得到的go条目主要与蛋白质定位或生物合成过程相关,都在癌症发生发展过程中的起到重要作用(图6f)。为了进一步验证这23个基因与食管癌之间的关系,我们比较了cancergenomeatlas收集的食管癌和正常样品的rna-seq数据。我们发现大多数这些基因的表达在癌症样品中上调(图6g)。根据这些发现,我们得出结论,cfdna的ngs可用于有效地表征染色质开放状态,并从表观遗传学的新观点识别癌症相关基因。
4.通过cfdna鉴定激活基因
为了进一步验证cfdna测序检测染色质状态的可靠性,我们将cfdna的ngs数据与食管癌细胞系的h3k27acchip-seq数据进行了比较,乙酰化水平可以作为染色质开放程度的重要证据。为了通过cfdna和两个食管癌细胞系的h3k27acchip-seq数据测序数据分别鉴定出染色质关闭和开放区域,对各组数据进行peakcalling。与cfdnareads分布相同(图4a),不同cfdna样品的染色质关闭区域的全基因组水平上的分布显示出极大的多样性(图8a)。比较由cfdna和chip-seq鉴定出的peak,重叠比例很低(图8b),表明cfdnangs和h3k27acchip-seq对染色质状态鉴定结果高度一致。对cfdnapeak分布的分析还表明,只有少部分的peak位于启动子区(图8c)。食管癌cfdna中鉴定出的位于启动子区的peak少于正常个体(图8c),表明食管癌患者的启动子区染色质开放程度高于正常人。进一步分析显示,食管癌cfdna富集得到的peak数量少于正常cfdna(图8d),这也表明在食管癌患者中染色质开放区域多于正常人。由于开放的染色质区域为基因的激活提供了机会,我们比较了癌症患者和正常人群中cfdnapeak的关联基因。结果显示,peak相关联的基因可分为三类(图8e),食管癌患者中特异表达基因比正常人特异表达基因(图8f)多约2.4倍,表明食管癌患者有更多基因被激活。go分析显示食管癌特异表达基因与各种刺激的应答密切相关,与由食管癌实体瘤组织中鉴定的差异表达基因富集得到的go条目类似(图9)。这表明通过cfdna的ngs数据能够鉴定在癌症过程中发挥关键作用的基因(图8g)。基于这些结果,我们得出结论,食管癌患者染色质开放程度更高,能够使更多食管癌特异性基因的激活。这些结果也与和染色质结构相关的基因启动子区开放程度高相一致,如ino80和whsc1(图6g)。
5.通过cfdna鉴定突变
靶向或全基因组水平测序检测突变是cfdna在nipt和液体活检中最广泛的应用。我们首先针对每个cfdna样品中的突变进行分析。通过比较不同类型cfdna的突变,发现在全基因组水平,不同样本间突变密度分布差异较大(图10a)。并且存在一些基因组区域,在所有样品中都具有较高或较低的突变密度(图10a)。作为美国食品和药物管理局(fda)在2017年授权的多基因检测panel(468基因),msk-impacttm可用于鉴定临床相关的体细胞突变,新的非编码突变,以及常见或罕见肿瘤中的突变特征。为了检验cfdnangs是否能检测到与临床相关的突变,我们将通过cfdna测序鉴定得到的存在突变的基因与msk-impactpanel收录的基因进行比较。发现术前患者特有的28个基因(bmpr1a、braf、cenpa、chek1、dcun1d1、epha3、erf、fgf3、gna11、grem1、inppl1、irf4、kdm5c、malt1、mitf、nf2、phox2b、pik3ca、rragc、rtel1、rxra、smad4、stk19、upf1、vhl、rptor)(图10b),表明这28个基因可能在食管癌的发生发展过程中起重要作用。go分析表明,这些基因与细胞死亡调节密切相关,表明这些基因在癌症发展过程发挥作用(图10c)。同时也富集得到了与结合或转录因子活性相关的go条目,这些功能也与基因表达调控存在密切联系,说明在食管癌患者中存在大量的基因表达(图10d),与图8f得出的结论一致。另外,通过对与msk-impactpanel重叠基因包含的突变进行注释,发现食管癌患者包含比正常人更多的非读码框改变的插入(图10d)。这种非读码框改变的插入来自于诸多典型的癌症相关基因,包括brca2,egfr,tnfaip3,alk,akt2,csf3r,dot1l,kdm5c,kmt2d,men1,prex2和ptch1(表6)。并且,这些基因都是食管癌特异性的。以上结果表明,cfdna可用于在液体活检中发现临床相关的体细胞突变。
表6.图10d中含有非读码框改变插入的基因
序列表
<110>东南大学
<120>一种高降解dna测序文库的构建方法及其应用
<160>25
<170>siposequencelisting1.0
<210>1
<211>36
<212>dna
<213>人工序列(artificialsequence)
<400>1
acactctttccctacacgacgctcttccgatctnnn36
<210>2
<211>33
<212>dna
<213>人工序列(artificialsequence)
<400>2
agatcggaagagcgtcgtgtagggaaagagtgt33
<210>3
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>3
gactggagttcagacgtgtgctcttccgatctacttgaagatgtgtataagagacagt58
<210>4
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>4
gactggagttcagacgtgtgctcttccgatctggctacagatgtgtataagagacagt58
<210>5
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>5
gactggagttcagacgtgtgctcttccgatctttaggcagatgtgtataagagacagt58
<210>6
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>6
gactggagttcagacgtgtgctcttccgatctcagatcagatgtgtataagagacagt58
<210>7
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>7
gactggagttcagacgtgtgctcttccgatcttgaccaagatgtgtataagagacagt58
<210>8
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>8
gactggagttcagacgtgtgctcttccgatctcgatgtagatgtgtataagagacagt58
<210>9
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>9
gactggagttcagacgtgtgctcttccgatctatcacgagatgtgtataagagacagt58
<210>10
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>10
gactggagttcagacgtgtgctcttccgatctcttgtaagatgtgtataagagacagt58
<210>11
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>11
gactggagttcagacgtgtgctcttccgatctacagtgagatgtgtataagagacagt58
<210>12
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>12
gactggagttcagacgtgtgctcttccgatcttagcttagatgtgtataagagacagt58
<210>13
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>13
gactggagttcagacgtgtgctcttccgatctgatcagagatgtgtataagagacagt58
<210>14
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>14
gactggagttcagacgtgtgctcttccgatctgccaatagatgtgtataagagacagt58
<210>15
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>15
gactggagttcagacgtgtgctcttccgatctagtcaaagatgtgtataagagacagt58
<210>16
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>16
gactggagttcagacgtgtgctcttccgatctgtccgcagatgtgtataagagacagt58
<210>17
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>17
gactggagttcagacgtgtgctcttccgatctccgtccagatgtgtataagagacagt58
<210>18
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>18
gactggagttcagacgtgtgctcttccgatctgtgaaaagatgtgtataagagacagt58
<210>19
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>19
gactggagttcagacgtgtgctcttccgatctattcctagatgtgtataagagacagt58
<210>20
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>20
gactggagttcagacgtgtgctcttccgatctagttccagatgtgtataagagacagt58
<210>21
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>21
gactggagttcagacgtgtgctcttccgatctactgatagatgtgtataagagacagt58
<210>22
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>22
gactggagttcagacgtgtgctcttccgatctgtggccagatgtgtataagagacagt58
<210>23
<211>19
<212>dna
<213>人工序列(artificialsequence)
<400>23
ctgtctcttatacacatct19
<210>24
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>24
aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatct58
<210>25
<211>66
<212>dna
<213>人工序列(artificialsequence)
<400>25
caagcagaagacggcatacgagattctgacatgtgactggagttcagacgtgtgctcttc60
cgatct66
- 还没有人留言评论。精彩留言会获得点赞!