构建多sgRNA表达载体的方法与流程
本发明涉及基因编辑领域。具体而言,本发明涉及构建多sgrna表达载体的方法。更具体而言,本发明涉及通过同源重组构建包含串联的靶向不同基因组位点的n个sgrna的表达载体的方法。
背景技术:
成簇的规律间隔的短回文重复序列(clusteredregularlyinterspacedshortpalindromicrepeats,crispr)及其相关蛋白9(crispr-associatedproteins9,cas9)在基础生物学研究、生物化学、农业、医药业等领域成为一种革命性的工具(barrangour,fremauxc,deveauh,etal.crisprprovidesacquiredresistanceagainstvirusesinprokaryotes[j].science,2007,315(5819):1709-1712.doudnaja,charpentiere.genomeediting.thenewfrontierofgenomeengineeringwithcrispr-cas9[j].science,2014,346(6213):1258096.hsupd,landeres,zhangf.developmentandapplicationsofcrispr-cas9forgenomeengineering[j].cell,2014,157(6):1262-1278.vanderoostj,westraer,jacksonrn,etal.unravellingthestructuralandmechanisticbasisofcrispr-cassystems[j].natrevmicrobiol,2014,12(7):479-492.barrangour,doudnaja.applicationsofcrisprtechnologiesinresearchandbeyond[j].natbiotechnol,2016,34(9):933-941.)。它设计简单、操作便捷且成本较低,可用来切割或结合特定的dna或rna序列,逐渐成为基因编辑、基因调控、基因治疗等技术的标准应用程序。2012年doudna等将crisprrna(crrna)与反式激活crrna(trans-activatingcrrna,tracrrna)连接并构建成单链向导rna(singleguiderna,sgrna)载体,证实与cas9一起可在体外切割dna片段。只需要改变sgrna中与目的基因互补的序列,就可以造成dna双链的断裂(double-strandbreak,dsb)(jinekm,chylinskik,fonfarai,etal.aprogrammabledual-rna-guideddnaendonucleaseinadaptivebacterialimmunity[j].science,2012,337(6096):816-821.)。断开的dna一般通过两条途径进行修复:主要是非同源末端连接(nonhomologousendjoining,nhej),造成断开位置的随机插入或缺失(insertionordeletion,indel)碱基(liebermr.themechanismofdouble-stranddnabreakrepairbythenonhomologousdnaend-joiningpathway[j].annurevbiochem,2010,79:181-211.)从而形成移码突变,进而造成基因敲除;另一条是同源重组修复途径(homologydirectedrepair,hdr),可利用带有同源臂的模板对切开位点进行特定修复(sanfilippoj,sungp,kleinh.mechanismofeukaryotichomologousrecombination[j].annurevbiochem,2008,77:229-257.),行使基因的插入、缺失、突变等功能。2013年fengzhang团队和church团队同时发表了,将crispr/cas9系统用于真核细胞中进行基因组编辑,在基因研究中具有里程碑式的意义(congl,ranfa,coxd,etal.multiplexgenomeengineeringusingcrispr/cassystems[j].science,2013,339(6121):819-823.malip,yangl,esveltkm,etal.rna-guidedhumangenomeengineeringviacas9[j].science,2013,339(6121):823-826.)。
sgrna作为crispr/cas9技术的重要组成部分,起引导cas9蛋白靶向目标dna的作用。为了同时对多个靶位点进行切割,就需要利用多个sgrna载体。然而,细胞中共转染多个质粒容易引起转染效率低下(wangh,yangh,shivalilacs,etal.one-stepgenerationofmicecarryingmutationsinmultiplegenesbycrispr/cas-mediatedgenomeengineering[j].cell,2013,153(4):910-918.)。sakuma等运用goldengate法将7个sgrna串联在一起构成all-in-one载体,对hek293t细胞进行转染之后发现,串联的sgrna载体同单独sgrna载体一样能够引起基因组编辑。但是其需要一系列用于串联的具有特定顺序的单独质粒,分别将20bp互补序列插入对应的单独质粒中,将u6-sgrna酶切下来之后连接在一起(sakumat,nishikawaa,kumes,etal.multiplexgenomeengineeringinhumancellsusingall-in-onecrispr/cas9vectorsystem[j].scirep,2014,4:5400.)。qi等以人工合成的方法串联了3个以trna为启动子的sgrna载体,证实了此系统在玉米中不仅能增加靶向基因的数目,还能提高突变效率(qiw,zhut,tianz,etal.high-efficiencycrispr/cas9multiplexgeneeditingusingtheglycinetrna-processingsystem-basedstrategyinmaize[j].bmcbiotechnol,2016,16(1):58.)。cao等以一步克隆法,构建多达6个sgrna的串联慢病毒载体,并证实了其可在细胞中起作用。但是其需要在引物两端设计多个互补的酶切粘性末端,引物设计步骤较为繁琐(caoj,wul,zhangsm,etal.aneasyandefficientinduciblecrispr/cas9platformwithimprovedspecificityformultiplegenetargeting[j].nucleicacidsres,2016,44(19):e149.)。xu等构建了单一启动子启动的,中间以自剪切核酶连接的多个sgrna串联载体,在小鼠及人的细胞中均能引起多基因编辑,但是第二个sgrna的合成效率明显低于第一个sgrna(xul,zhaol,gaoy,etal.empowermultiplexcellandtissue-specificcrispr-mediatedgenemanipulationwithself-cleavingribozymesandtrna[j].nucleicacidsres,2017,45(5):e28.)。yin等以in-fusion法构建含有4个sgrna串联的、各以u6启动子启动转录的aav载体,包装病毒后感染hiv-1模型小鼠,发现能有效清除hiv前病毒(yinc,zhangt,qux,etal.invivoexcisionofhiv-1provirusbysacas9andmultiplexsingle-guidernasinanimalmodels[j].molther,2017,25(5):1168-1186.),但是需要先构建2个sgrna串联载体,在此基础上再构建4个sgrna串联的重组载体。
本领域仍然需要一种构建串联sgrna的重组载体的快速有效的方法,从而提高提高试验效率,节约试验成本。
附图说明
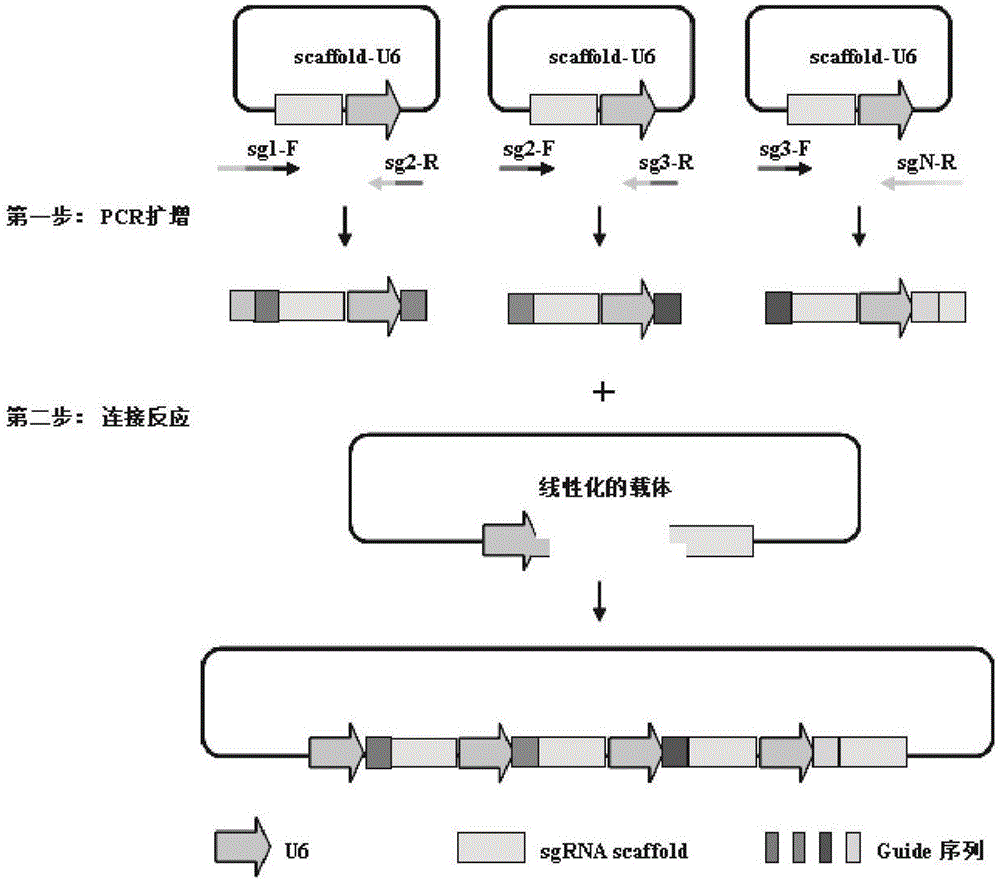
图1示出两步连接法构建串联sgrna载体示意图。首先,从scaffold-u6载体中以pcr的方法扩增得到两端带有同源臂片段(第一步);同线性化的载体进行连接(第二步)即可得到sgrna串联重组载体。u6:u6启动子。
图2示出t7ei检测串联sgrna重组载体在nih3t3细胞中基因编辑结果。图2a:mmstn-sgrna1位点t7ei法检测结果;图2:mmstn-sgrna2位点t7ei法检测结果;图2c:mtyr-sgrna3位点t7ei法检测结果;图2d:mrosa26-sgrna2位点t7ei法检测结果。su:以u6为启动子的一个sgrna组;u4:串联4个sgrna重组载体;m:50bpdnaladder;c-:不含向导序列的空载体;红色箭头指示切开的目的片段。
图3示出串联sgrna重组载体酶切鉴定结果。图3a:酶切位点示意图,nsgrna表示串联不同数量的sgrna;图3b:以kpni和noti双酶切鉴定串联不同数量的sgrna重组载体结果。1sg-6sg:串联1-6个sgrna的重组载体;c-:不含向导序列的空载体;m:dl10000;“*”代表切出正确的目的条带;右侧碱基长度代表酶切的目的片段大小。
发明详述
除非另有指示或定义,否则所有所用术语均具有本领域中的通常含义,该含义将为本领域技术人员所了解。参考例如标准手册,如sambrook等人,“molecularcloning:alaboratorymanual”(第2版),第1-3卷,coldspringharborlaboratorypress(1989);lewin,“genesiv”,oxforduniversitypress,newyork,(1990);及roitt等人,“immunology”(第2版),gowermedicalpublishing,london,newyork(1989),以及本文中引用的一般现有技术;此外,除非另有说明,否则未具体详述的所有方法、步骤、技术及操作均可以且已经以本身已知的方式进行,该方式将为本领域技术人员所了解。亦参考例如标准手册、上述一般现有技术及其中引用的其他参考文献。
在说明书中和权利要求中所使用的,指不同的结构或方法步骤的序数指示,比如第一、第二和第三,不应该被解释为指示任何具体的结构或步骤、或者这种结构或步骤的任何特定顺序或构型。在本文中描述的所有方法可以以任何合适的顺序进行,除本文中另有指示,或者明显与上下文矛盾。
本发明提供了一种构建包含串联的靶向不同基因组位点的n个sgrna编码序列的表达载体的方法,其包括以下步骤:
1)提供如下的n-1个dna片段:
第1个片段:g1-s-p-g2;……第n-1个片段:gn-1-s-p-gn,
其中g1,……gn代表n个分别靶向不同基因组位点的sgrna向导序列,s代表sgrna的框架序列,p代表驱动sgrna表达的启动子,n是选自2-10的整数,
其中所述第1个片段的5′端还包含所述驱动sgrna表达的启动子p或其部分序列,所述第n-1个片段的3′端还包含所述sgrna的框架序列s或其部分序列;
2)提供线性化的表达载体,其中所述线性化的表达载体的一端具有所述驱动sgrna表达的启动子p或其部分序列,另一端具有所述sgrna的框架序列s或其部分序列;
3)将所述n-1个片段与所述线性化的表达载体混合,在合适的条件下进行同源重组,
由此产生包含串联的靶向不同基因组位点的n个sgrna编码序列的表达载体。
在一些实施方案中,所述线性化的表达载体的一端具有所述驱动sgrna表达的启动子p的部分序列与第1个片段的5′端的所述驱动sgrna表达的启动子p的部分序列重叠,从而能进行同源重组。在一些实施方案中,所述线性化的表达载体的一端的所述sgrna的框架序列s的部分序列与第1个片段的3′端的所述sgrna的框架序列s的部分序列重叠,从而能进行同源重组。
根据本发明的方法,所述n-1个dna片段可以便利地通过pcr等手段扩增得到或可以直接化学合成获得,并且通过直接使用不同的sgrna向导序列作为同源重组中的同源臂,可以在一个反应内将n个启动子-sgrna序列串联克隆进表达载体。
例如,如果n是3,则可产生两个dna片段:第1个片段:g1-s-p-g2;…和第2个片段:g2-s-p-g3。其中g2可以作为第1个片段和第2个片段之间同源重组的同源臂。
例如,如果n是4,则可产生3个dna片段:第1个片段:g1-s-p-g2;第2个片段:g2-s-p-g3;和第3个片段:g3-s-p-g4。其中g2可以作为第1个片段和第2个片段之间同源重组的同源臂,g3可以作为第2个片段和第3个片段之间同源重组的同源臂。由于g1、g2、g3和g4具有不同的序列,因此可以在同一同源重组反应中实现这些片段的特定顺序的串联。当n是更大的整数时,以此类推。
因此,本发明的方法可以大大缩短工作时间并节约实验成本。
如本文所用,“sgrna”和“单向导rna”可互换使用,指的是crispr基因组编辑系统中能够与crispr核酸酶形成复合物并由于与靶序列具有一定互补性而能够将所述复合物靶向靶序列的rna分子。例如,在基于cas9的基因编辑系统中,grna通常由部分互补形成复合物的crrna和tracrrna分子构成,其中crrna包含与靶序列具有足够互补性以便与该靶序列杂交并且指导crispr复合物(cas9+crrna+tracrrna)与该靶序列序列特异性地结合的序列。然而,本领域已知可以设计单向导rna(sgrna),其同时包含crrna和tracrrna的特征。基于所使用的cas9多肽和待编辑的靶序列设计合适的sgrna序列属于本领域技术人员的能力范围内。
在本发明中,“向导序列”和“guide序列”可以互换地使用,是指sgrna中与目标基因的靶标序列互补的部分。sgrna与靶标序列互补的部分的长度可以在10个核苷酸、13个核苷酸、15个核苷酸、18个核苷酸、20个核苷酸、22个核苷酸或24个核苷酸之间,或者在10至30之间的任意数目的核苷酸。sgrna与靶标序列互补的部分应该能够与在靶标链中的序列杂交,并且最佳地完全与靶标序列互补。
sgrna的框架序列是指sgrna中除了向导序列之外的序列,其主要负责与crispr核酸酶相互作用。sgrna的框架序列根据所使用的不同crispr核酸酶而有所不同。在一些实施方案中,sgrna的框架序列是cas9核酸酶的sgrna的框架序列。
驱动sgrna表达的启动子可以是rna聚合酶iii启动子,例如u6启动子。
在一些实施方案中,所述线性化的表达载体还含有另一启动子,以及与所述另一启动子可操作地连接的编码crispr核酸酶的多核苷酸。任选地,所述编码crispr核酸酶的多核苷酸还与调控基因表达的其他元件可操作地连接,例如终止子等。
在一些实施方案中,所述另一启动子是rna聚合酶ii启动子,包括但不限于ef1α启动子、cmv启动子、cba启动子、hsynapsin启动子、hsv-tk启动子、sv40早期启动子和lsp启动子,优选cmv启动子。
如本文所用,术语“crispr核酸酶”通常指在天然存在的crispr系统中存在的核酸酶,以及其修饰形式、其变体(包括切口酶突变体)、或其催化活性片段。crispr核酸酶可以通过与向导rna一起相互作用来识别、结合和/或切割靶核酸结构。该术语涵盖基于crispr系统的能够在细胞内实现基因编辑的任何核酸酶或其功能性变体。本发明的crispr核酸酶例如可以选自cas3、cas8a、cas5、cas8b、cas8c、cas10d、cse1、cse2、csy1、csy2、csy3、gsu0054、cas10、csm2、cmr5、cas10、csx11、csx10、csf1、cas9、csn2、cas4、cpf1、c2c1、c2c3或c2c2蛋白,或这些核酸酶的功能性变体。
在一些优选实施方案中,所述crispr核酸酶是cas9。在一些实施方案中,所述cas9为金黄色葡萄球菌(staphylococcusaureus)cas9或其变体。
不同cas9多肽依赖于不同的识别位点或pam,sacas9的pam是5′-nngrrt-3′,其中n是任意核苷酸,r是嘌呤。不同cas9多肽具有不同的sgrna支架序列,形成单向导rna的3′部分。sgrna的靶标序列特异性的5′部分(即向导序列)的长度也同样在cas9核酸酶酶间不同,sacas9使用18至24个核苷酸靶标序列。所述cas9核酸酶变体的实例包括但不限于cas9核酸酶的高特异性变体,例如pct/us2016/049147、pct/us2016/020756等描述的sacas9核酸酶变体。本发明可用的其他一些“cas9多肽”可见于例如http://www.addgene.org/crispr/guide/。
如本文所用,术语“可操作地连接”描述调控元件和基因或其编码区之间的连接。即,通常基因表达位于某种调控元件的控制下,例如不限于组成型或诱导型启动子、组织特异性调控元件和增强子。称基因或编码区域与调控元件“可操作地连接”,意思是基因或编码区域受调控元件的控制或影响。在本发明中,调控元件包括启动子、增强子、反式激活因子等。
在一些实施方案中,所述线性化的表达载体为线性化的质粒载体。在一些实施方案中,所述质粒载体用于病毒载体如腺相关病毒(aav)的包装。
在一些具体实施方案中,所述质粒载体衍生自px601载体。
如本文所用,“合适的条件”是指能够使各个片段和线性化的表达载体通过同源重组而按所需顺序连接的条件。例如,可以使用clonexpressmultisonestepcloning试剂盒,以及合适的片段的量实现所述同源重组。本领域技术人员可以合适地确定所需要的各个片段的量。
在另一方面,本发明还涉及通过本发明的方法制备的包含串联的靶向不同基因组位点的n个sgrna编码序列的表达载体。所述载体特别适合于在细胞中同时对多个基因组位点进行crispr介导的基因编辑。
在另一方面,本发明提供了一种试剂盒,其包含本发明的表达载体。
试剂盒一般包括表明试剂盒内容物的预期用途和/或使用方法的标签。术语标签包括在试剂盒上或与试剂盒一起提供的或以其他方式随试剂盒提供的任何书面的或记录的材料。
实施例
在此描述的实施例是用于说明的目的,并不意在限制本发明的范围,本领域技术人员可以根据本发明的精神和教导对具体步骤进行修改。除非另有规定或从内容明显看出,否则所记载的与一些实施方案有关的任何特征可以与任何其他实施方案来结合使用。
实施例1构建支架-u6重组载体
首先分别扩增支架序列、带有支架同源序列的u6序列,再以重叠延伸pcr法将其组合,并将其与pmd19-t连接,测序正确后,命名为支架-u6。
以px601质粒(购自addgene,质粒编号61591)为模板(10ng/体系),以引物:
scaf-f:5′-gttttagtactctggaaaca-3′和
scaf-r:5′-aaaaaaatctcgccaacaagttg-3′;
scaf-u6-f:
5′-caacttgttggcgagatttttttgagggcctatttcccatgatt-3′和u6-r:5′-ggtgtttcgtcctttccaca-3′
分别用q5dna聚合酶进行pcr扩增,扩增结束后,各加入dpni1μl,37℃孵育30min消化质粒模板结束后,1%琼脂糖凝胶120v电泳25min,并胶回收所得目的条带。
以所得胶回收产物1μl为模板(各10ng),以scaf-f和u6-r为引物,用q5高保真dna聚合酶进行pcr扩增。pcr结束后,各加入rtaq0.5μl,37℃孵育30min。孵育结束后,1%琼脂糖凝胶120v电泳25min,并胶回收所得目的条带,得到支架-u6扩增片段。
pmd19-t是本领域技术人员已知的一种t载体,其是一种高效克隆pcr产物(ta克隆)的专用质粒载体,为线性化载体,无需酶切可直接与具有a末端的pcr产物连接,属于非定向克隆。将所得支架-u6扩增片段按照生产商的说明书连接pmd19-t,4℃连接过夜。转化进入top10感受态细胞,预先在lb(amp+,100μg/ml)板中加入iptg10μl和x-gal30μl,37℃培养过夜后,挑取白色菌落摇菌并送测序鉴定,鉴定正确的样品命名为支架-u6。
实施例2构建靶向多个靶标的sgrna串联重组载体
以串联4个sgrna为例,本发明以3个小鼠内源基因的4个sgrna位点,依次为:mmstn-sgrna1、mmstn-sgrna2、mtyr-sgrna3、mrosa26-sgrna2,以px601为载体骨架将其串联,构建sgrna串联重组载体。
如图1所示设计引物,其中,sg1-f引物(5′-3′)包括20bp的u6启动子3′端序列,加上第一个sgrna的向导序列(约22bp),再加上18bp的支架5′端序列;sgn-r引物(5′-3′)包括20bp反向的支架5′端序列,加上反向的第n个sgrna的向导序列(约22bp),再加上18bp反向的u6启动子3′端序列;其余sgrna引物序列除了带有特定的向导序列,正向引物还带有18bp的支架5′端序列,反向引物则带有18bp的u6启动子3′端序列。引物序列表1所示:
表1
以支架-u6质粒为模板(10ng/体系),以引物:sg1-f/sg2-r、sg2-f/sg3-r、sg3-f/sg4-r进行pcr扩增,扩增结束后,用dpni消化质粒模板,方法同实施例1,获得含有同源臂的扩增片段。
以bsai将px601进行酶切,在pcr仪中,37℃孵育30min。结束后,1%琼脂糖凝胶120v电泳25min,并胶回收所得目的条带,获得px601经bsai酶切片段。
将含有同源臂的扩增片段和px601经bsai酶切片段按以下公式计算用量:
插入的dna片段用量(μl)=20ng/yng/μl
线性化载体用量(μl)=(xbp×0.02)/yng/μl
将胶回收产物用clonexpressmultisonestepcloning试剂盒(购自南京诺唯赞,批号7e002g6)进行连接,参照该说明书操作,连接体系如下:
37℃孵育30min后,立即冰中>5min后进行转化进入xl10-gold感受态细胞,涂lb(amp+,100μg/ml)板并37℃孵育过夜。
最后,以引物:sg1-test-f:5′-gaaacaatcattaccatgccta-3′和sg4-test-r:5′-gcccatcttctagaaagactgc-3′,进行菌液pcr鉴定,将菌液pcr鉴定阳性样品送测序,测序正确样品进行中提质粒,获得4个sgrna串联重组载体。
实施例3检测基因编辑效果
3.1细胞转染
转染前一天,nih3t3细胞按每孔5.2×105个细胞/孔轻轻加入6孔板中。将100μldmem培养基、4μg质粒和8μlturbofect(购自thermoscientific,批号00448764)充分混匀后,室温静置15-20min,按所需量加入各孔中后,轻轻晃动6孔板混匀。放入细胞培养箱中,37℃,5%co2条件下培养。转染8h后,吸弃废液,各加3ml10%胎牛血清培养液继续培养。
转染72h后,用细胞/细菌/酵母基因组小量提取试剂盒(上海莱枫,批号j0301),按照生产商说明书提取基因组dna。
3.2t7核酸内切酶i法检测基因编辑效果
以200ng基因组dna为模板,以如表2所示的各sgrna位点检测引物,用q5高保证dna聚合酶扩增,1%琼脂糖凝胶电泳,并胶回收所得目的条带。
表2
用q5高保证dna聚合酶扩增,1%琼脂糖凝胶电泳,并胶回收所得目的条带。
将200ng所得pcr产物分别加入含有nebuffer2的管中,并设置未用质粒转染的nih3t3细胞基因组为阴性对照(c-)。在pcr仪中进行加热变性、退火复性处理,结束后,每管加入0.4μlt7核酸内切酶i,37℃孵育1h。
各样品中加入2μl6×loaddingbuffer,page变性胶电泳110v,50min。电泳结束后,sybrgreeni染色1h,凝胶成像系统拍照(图2a-d)。
t7核酸内切酶i识别并切割带有切割位点的产物。箭头所指为含有突变的dna。以imagej软件分析同泳道内各条带(a为pcr扩增片段,b、c为t7核酸内切酶i酶切出的目的条带)灰度值,通过公式(b+c)/(a+b+c)计算indel比例。
从图2可知,串联4个sgrna重组载体的编辑效率约等于或大于只有一个u6启动子的载体。
3.3双酶切鉴定串联sgrna重组载体
本发明构建串联1-6个sgrna重组载体,并以kpni和noti双酶切进行鉴定,在pcr仪中,37℃孵育30min。结束后,1%琼脂糖凝胶120v电泳25min,凝胶成像系统拍照(图3)。
如图3所示,根据本发明的方法成功构建串联1-6个sgrna的重组载体。所述载体均能够成功地靶向靶标基因并进行切割,从而缩短工作时间并节约试验成本。
- 还没有人留言评论。精彩留言会获得点赞!