一种利用基因编辑技术敲入终止子实现可转录元件敲除的方法与流程
本发明涉及生物技术领域,具体涉及一种更高效,应用范围更广泛(包括非编码rna)的基于基因编辑技术的基因敲除方法。
背景技术:
基因敲除(knockout)是一种遗传工程技术,通过在基因组层面对生物的遗传物质改变,令特定的基因丧失功能,在研究实践中,往往通过目的基因的敲除观察和检测对生物体造成的影响,从而推测出该基因功能。是功能基因组研究的重要工具,也为一些疾病的治疗,工具生物的改进提供强大的技术支持。
传统的基因敲除建立在胚胎干细胞技术和基因同源重组技术基础上的一种分子生物学技术,效率比较低。基因编辑对生物医学研究的影响不亚于30年前的pcr。基因编辑技术的出现为我们敲除基因提供了更好的策略。近年来,锌指核酸酶(zfn)、转录激活因子样效应子(talen)和规律成簇的间隔短回文重复(crispr)已经掀起了一浪又一浪的基因编辑高潮。尤其是crispr技术,由于其简单有效,被认为是近年出现的革命性的基因编辑技术。该技术可以快速,容易的实现对目标dna序列在精确的位点进行编辑,包括突变,修改,插入等改变,因此,基于crispr技术的基因敲除目前有广泛的应用。crispr(clusteredregularlyinterspacedshortpalindromicrepeats)即成簇规律间隔短回文重复,是存在于细菌基因组中的dna序列,cas(crisprassociated)即crispr相关基因,有多种类型,cas9是其中一种。在以cas9作为核酸内切酶的情况下,通过向细胞内导入grna和cas9蛋白实现对目标dna的切割。grna是一个经过特殊设计的向导rna,可以识别并结合靶基因dna序列,cas9蛋白是一个酶,可以与grna以及其识别的dna结合,将目的dna序列切断,再利用细胞dna的损伤修复机制,实现对dna在断裂位点及附近进行突变,插入,替换等等修改,实现基因编辑的目的。因此crispr系统常被比作一把可定点切割dna的“魔剪”。(参见文献:mali,p.,yang,l.,esvelt,k.m.,aach,j.,guell,m.,dicarlo,j.e.,norville,j.e.,andchurch,g.m.(2013).rna-guidedhumangenomeengineeringviacas9.science339,823-826)。
在其最常应用的场景中,crispr系统被用于蛋白编码基因的敲除。因为只需要利用crispr系统在蛋白编码基因的编码区(sequencecodingforaminoacidsinprotein,cds)起始位置“剪一刀”,即产生一个dna双链断裂(doublestrandbreak,dsb),此时dna主要通过非同源末端连接(non-homologousendjoining-nhej)的方式修复,会导致切割位点的插入或缺失突变(indel),因此,该基因在表达成蛋白时,大概率会出现框移突变,使得正确的蛋白不能表达,从而实现基因的功能敲除。但是,crispr介导的dsb有一定的效率,实际操作中需要把导入crispr个元素的细胞分成单细胞培养,一个一个挑克隆进行下一步的鉴定,工作量非常大。而且,如果该策略只能在cds区域切割dna才有效,而设计grna必须满足很多条件,比如有pam(protospaceradjacentmotif)序列,无脱靶位点等,使得有的基因的cds区并不能设计grna,使得该策略的应用范围降低。
另外,目前利用产生一个dna双链断裂(doublestrandbreak,dsb)的该策略不能敲除非编码rna,因为非编码rna不存在框移突变的问题,局部的突变可能完全不影响功能。因此非编码rna的现有敲除策略一般采用“剪两刀”,即在非编码rna的上下游各产生一个dsb,将非编码rna所在基因序列删除。但这依然有效率低,挑单克隆更困难的问题,而且,如果非编码rna所在基因序列是一个增强子或其他关键元件,其丢失必然导致严重的实验偏差。
目前关于一种更高效,应用范围更广泛,能够应用于非编码rna的基于基因编辑技术的基因敲除方法还未见报道。
技术实现要素:
基于现有技术的缺陷,能否从更源头寻找其他敲除编码和非编码基因的方法呢?我们知道,不管是编码基因和还是非编码rna,其终末功能物质都要来自dna的转录,而dna的转录终止是受到序列信息调控的,转录过程中提供终止信号的dna序列就是终止子(terminator)。
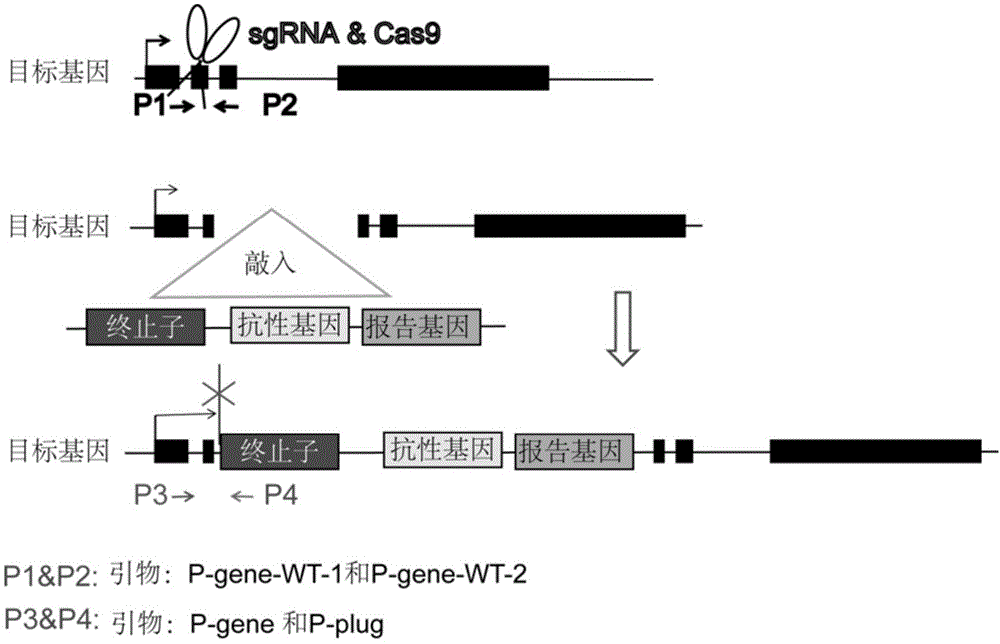
据此,本发明提供了一种新的方法,即利用基因编辑技术(如crispr技术),在目的基因的转录起始位点的下游,在不利用同源臂的情况下,敲入转录终止dna序列(终止子),实现目的基因的功能敲除(见图1)。该方法可以通过“剪一刀”既可以敲除蛋白编码基因,且设计在内含子区也有效,增加可被敲除蛋白编码基因的范围,还可以敲除非编码rna的母基因。而且,本发明的包含终止子的dna载体可以包含药物筛选基因(比如嘌呤霉素抗性基因),使得成功敲入的细胞可以被筛选出来,大大增加单克隆挑选的效率。最后,本发明设计利用非同源末端连接(nhej)途径敲入,因此在终止子两端不需要为目的基因设计同源臂,使得包含终止子的dna载体具有通用性。本方法可能有较大的实用价值。
目前尚无文献报道利用非同源末端连接(nhej)途径敲入终止子实现蛋白编码基因和非编码rna母基因的策略。
本发明的目的在于提供一种基于基因编辑技术(如crispr技术)敲入终止子实现可转录元件的方法。所述可转录元件包括蛋白编码基因及非编码基因。本发明的敲除方法相较目前已有基因敲除方法,策略不同,效率更高,应用范围更广泛(包括非编码rna)。
为了实现上述目的,本发明提供一种利用基因编辑技术敲入终止子实现可转录元件敲除的方法,包括如下步骤:
(a)构建包含转录终止序列的dna序列,所述的包含转录终止序列的dna序列不含所要敲除的可转录元件的同源序列;
(b)利用基因编辑技术在目的基因的转录起始位点下游敲入所述的包含转录终止序列的dna序列。
进一步的,所述的可转录元件包括蛋白编码基因和非编码基因。所述的非编码基因包括长链非编码rna、microrna、snorna等由基因组dna被转录所产生的分子。
进一步的,所述的基因编辑技术指能够让人类对目标基因进行“编辑”,实现对特定dna碱基或片段的敲除、加入等修改的技术,例如crispr技术、talen技术、zfn技术等。
进一步的,所述的包含转录终止序列的dna序列中还包括抗性基因元件。所述的抗性基因包括neomycin抗性基因,hygromycinb抗性基因,嘌呤霉素抗性基因,潮霉素抗性基因,杀稻瘟菌素抗性基因等。
进一步的,所述的包含转录终止序列的dna序列中还包括报告基因。所述的报告基因为荧光基因等,如egfp(增强绿色荧光蛋白,enhancedgreenfluorescentprotein)。
进一步的,所述的转录终止序列是dna上被识别后引起rna转录终止的dna序列,例如:svc0poly(a)signal,beta-globinpoly(a)signal,hsvtkpoly(a)signal,bghpoly(a)signal,tk_pa_terminator,连续4个以上的t,依赖或者不依赖于蛋白质辅因子的转录终止子等。
进一步的,所述的步骤b中在目的基因的转录起始位点下游敲入所述的包含转录终止序列的dna序列,敲入位点尽量靠近目的基因的转录起始位点(tss),并避免出现脱靶位点。
进一步的,本发明所述的一种利用基因编辑技术敲入终止子实现可转录元件敲除的方法,包括如下步骤:
步骤a:构建包含转录终止序列的dna序列,所述的包含转录终止序列的dna序列不含所要敲除的可转录元件的同源序列;
步骤b:根据所要敲除的目的基因或非编码rna母基因,设计并构建针对目的基因和包含转录终止序列的dna序列的基因编辑技术元件;
步骤c:将构建的基因编辑技术元件导入目标细胞,在目的基因的转录起始位点下游敲入所述的包含转录终止序列的dna序列,实现对于目的基因的敲除。
更进一步的,本发明所述的一种利用基因编辑技术敲入终止子实现可转录元件敲除的方法,具体包括如下步骤:
步骤一:构建包含转录终止序列(终止子)的dna序列(不需要同源臂,即所述的包含转录终止序列的dna序列不含所要敲除的可转录元件的同源序列)。
所述的“包含转录终止序列的dna序列”通常是质粒,也可由人工合成的dna替代,为描述生动,本发明将所述的“包含转录终止序列的dna序列”比喻成“塞子,plug”,可以阻塞转录,让转录过程停于“塞子”处,“塞子”下游的序列不被转录。本发明的方法建议在“塞子”里加入抗性基因元件(比如neomycin抗性基因,嘌呤霉素抗性基因,潮霉素抗性基因,杀稻瘟菌素抗性基因等),根据实际需要设计构建,有抗性基因可以筛选插入“塞子”的细胞,增加后续单克隆挑选的效率,根据实际需要,也可以不加抗性基因,但效率会降低。除此以外,在“塞子”上,还可以加入荧光基因等其他报告基因,增加操作者对敲除过程的把握度。
步骤二:选择干预目标种属,确定想要敲除的目的基因或非编码rna母基因,设计基因切断的基因编辑技术元件。
若利用crispr技术,设计识别grna:grna-gene。设计切断包含转录终止序列(终止子)的dna序列的grna:grna-plug。若选择talen(转录激活因子类感受器核酸酶)技术,设计特异性talen-gene和talen-plug元件。
注:经过本发明的实验,我们发现将拟敲入的“塞子”环化,再在细胞内利用基因编辑元件如grna-plug线性化可以极大的提高敲入的效率,该过程依赖于nhej修复途径,体内线下化“塞子”是本发明的一个优化步骤,不进行该步骤也可以实现目的基因敲除,也属于本发明内容的一部分。
步骤三:分别构建针对目的基因和“塞子”的基因编辑元件。
若选择crispr技术中的cas9相关技术,可以构建grna-gene,grna-plug表达载体及cas9表达载体。若选择talen技术,构建特异性talen-gene和talen-plug表达载体。(这些表达载体可以是质粒,也可以包装成病毒,也可直接是rna,形式可以很多,也可以将这些元件体外合成,后续直接将rna和蛋白质导入细胞)。
步骤四:将分别构建的基因编辑元件导入目标细胞。
若选择crispr-cas9技术,将grna-gene,grna-plug,以及cas9导入目标细胞。若选择talen技术,将talen-gene和talen-plug导入目标细胞。若选择其他基因编辑技术,将相应元件导入细胞。
进一步的,本发明的方法中还可以包括以下步骤五、六、七:
步骤五:加药筛选“塞子”整合入细胞基因组的细胞。
对于在“塞子”里加入了抗性基因元件和/或报告基因的,根据“塞子”的抗药性加入药物筛选成功插入基因组的细胞,该步骤可以大大提高后续单细胞可能挑选鉴定的效率。
本发明的方法建议在“塞子”里加入抗性基因元件(比如neomycin抗性基因,嘌呤霉素抗性基因,潮霉素抗性基因,杀稻瘟菌素抗性基因等),根据实际需要设计构建,有抗性基因可以筛选插入“塞子”的细胞,增加后续单克隆挑选的效率,根据实际需要,也可以不加抗性基因,但效率会降低。除此以外,在“塞子”上,还可以加入荧光基因等其他报告基因,增加操作者对敲除过程的把握度。
步骤六:鉴定“塞子”整合入目的基因设计位置。
根据终止子被敲入的位置,可设计pcr反应引物,pcr扩增并测序扩增产物,用于鉴定是否有细胞的基因组上相应位置成功敲入“塞子”(图1)。
步骤七:克隆化成功敲除目的基因的细胞系并鉴定。
上述六步已经得到部分细胞目的基因被敲除的细胞混合物了,根据需要还可进行单细胞克隆的挑选,并进一步鉴定目的基因被敲除,得到目的基因敲除的细胞株。
所述的步骤一,构建包含转录终止序列(终止子)的dna序列(即“塞子”),具体为:
利用分子克隆技术构建,或者委托基因合成公司直接合成一个包含转录终止序列(终止子)的载体,所述载体为dna分子,所述转录终止序列可根据不同的细胞类型和敲除基因类型进行选择,比如真核生物中针对rna聚合酶ii的转录终止可以选择svc0poly(a)signal,beta-globinpoly(a)signal,hsvtkpoly(a)signal,bghpoly(a)signal,tk_pa_terminator等等,针对rna聚合酶iii可以选择连续4个以上的t。比如原核细胞中选择依赖或者不依赖于蛋白质辅因子(如:ρ因子)的转录终止子。本发明的方法建议在塞子里加入抗性基因元件(比如neomycin抗性基因,嘌呤霉素抗性基因,潮霉素抗性基因,杀稻瘟菌素抗性基因等),根据实际需要设计构建,有抗性基因可以筛选插入“塞子”的细胞,增加后续单克隆挑选的效率,根据实际需要,也可以不加抗性基因,但效率会降低。除此以外,在该载体上,还可以加入荧光基因等其他报告基因,增加操作者对敲除过程的把握度。也可以是市面上已有的包含转录终止序列的载体,设计进入细胞后的载体断点,使得已有转录终止序列发挥终止内源要靶向的基因的作用即可。敲入终止子不依赖同源重组敲除基因策略见图1。
在本发明的一个优选实施方式中,选择了tk_pa_terminator(其侧翼有tttt,既可以终止rna聚合酶ii的转录,又可以终止rna聚合酶iii的转录,其序列如seqidno.1所示)作为转录蛋白编码基因和长链非编码rna的转录终止序列。
在本发明的一个优选实施方式中,选择neomycin抗性基因作为筛选基因。
在本发明的另一个优选实施方式中,选择hygromycinb抗性基因作为筛选基因。
在本发明的一个优选实施方式中,选择egfp(增强绿色荧光蛋白,enhancedgreenfluorescentprotein)作为报告基因。
在本发明的一个优选实施方式中,所述的基因编辑技术为crispr-cas9技术,所述的包含转录终止序列的dna序列为包含转录终止序列tk_pa_terminator、含有neomycin抗性基因,并表达egfp的序列。
在本发明的一个优选实施方式中,所述的基因编辑技术为crispr-cas9技术,所述的包含转录终止序列的dna序列为包含转录终止序列tk_pa_terminator、含有hygromycinb抗性基因的序列。
所述的步骤二:选择干预目标种属,确定想要敲除的目的基因或非编码rna母基因,设计基因切断的基因编辑技术元件。具体为:
在目的基因的转录起始位点(tss)之后设计切点,尽量靠近tss。如果利用crispr技术,其所识别的染色体位点上满足pam序列要求,并避免出现脱靶位点。
特别的,如果利用crispr-cas9进行切割,设计的方式采用(n)19ngg的序列模式的,n为a、t、c或g,下标19表示n的个数;具体设计方法可参考以下网址:
http://www.addgene.org/static/cms/files/hcrispr_grna_synthesis.pdf。
针对包含转录终止子的“塞子”的线性化的切割元件设计也要遵循并避免出现脱靶位点的原则,并且切点附近就是转录终止子,使得“塞子”被插入目的基因切割位置后,终止子正好位于目的基因tss下游。
在本发明的一个优选实施方式中,我们选择了crispr-cas9技术作为基因编辑技术,选择了哺乳动物中的人类的基因组作为设计干预目标,长链非编码rnattty15作为拟敲除的非编码rna,设计识别grna:grna-ttty15,其识别位点序列如下,tcaggcaatccagccgcccggg(seqidno.2),其中ggg为pam序列。cas9表达载体为商品化的质粒和病毒。
在本发明的一个优选实施方式中,我们选择了crispr-cas9技术作为基因编辑技术,选择了哺乳动物中的人类的基因组作为设计干预目标,蛋白编码基因mms22l作为拟敲除的蛋白编码基因,设计识别grna:grna-mms22l,其识别位点序列如下,gcttaagggctccgctgcagagg(seqidno.3),其中agg为pam序列。cas9表达载体为商品化的质粒和病毒。
在本发明的一个优选实施方式中,我们选择了crispr-cas9技术作为基因编辑技术,选择了哺乳动物中的人类的基因组作为设计干预目标,mir-155作为拟敲除的microrna,设计识别grna:grna-mir-155,其识别位点序列如下,gttaatgctaatcgtgatagggg(seqidno.4),其中ggg为pam序列。cas9表达载体为商品化的质粒和病毒。
所述的步骤三:分别构建针对目的基因和“塞子”的基因编辑元件。具体为:
利用分子克隆技术构建或委托基因合成公司直接合成基因编辑元件表达载体,这些元件分别针对目的基因和“塞子”,使得目的基因最终在细胞内被精准切开,拟插入的含有终止子的“塞子”也被切开线性化。
若选择crispr技术中的cas9相关技术,可以构建grna-gene,grna-plug表达载体及cas9表达载体。若选择talen技术,构建特异性talen-gene和talen-plug表达载体。(这些载体可以是质粒,也可以包装成病毒,也可直接是rna,形式可以很多,也可以将这些元件体外合成,后续直接将rna和蛋白质导入细胞)。
在本发明的一个优选实施方式中,我们选择了crispr-cas9技术作为基因编辑技术,grna-ttty15和grna-plug的表达元件被分别构建到慢病毒骨架载体plko-bsd中(这两个质粒可以被包装成慢病毒)。cas9表达载体为商品化的质粒cas9表达载体购自addgene(plasmidid号:41815)。
在本发明的一个优选实施方式中,我们选择了crispr-cas9技术作为基因编辑技术,grna-mms22l和grna-plug的表达元件被分别构建到慢病毒骨架载体plko-bsd中(这两个质粒可以被包装成慢病毒)。cas9表达载体为商品化的质粒cas9表达载体购自addgene(plasmidid号:41815)。
在本发明的一个优选实施方式中,我们选择了crispr-cas9技术作为基因编辑技术,grna-mir-155和grna-plug的表达元件被分别构建到慢病毒骨架载体plko-bsd中(这两个质粒可以被包装成慢病毒)。cas9表达载体为商品化的质粒cas9表达载体购自addgene(plasmidid号:41815)。
所述的步骤四:将分别构建基因编辑元件导入目标细胞。具体步骤为:
选取需要敲除的目的细胞,将细胞培养于培养基中,状态良好的情况下,通过脂质体等方式将构建进入质粒的针对目的基因和“塞子”的基因编辑元件转染入细胞,或者利用病毒将构建进入质粒的针对目的基因和“塞子”的基因编辑元件感染入细胞。如果选取的靶细胞较大,如卵细胞,还可以将基因编辑元件显微注射到靶细胞。
在本发明的一个优选实施方式中,我们将grna-ttty15,grna-plug以及cas9的表达质粒通过lipofectamine3000转染进入前列腺癌细胞系du145中。
在本发明的一个优选实施方式中,我们将grna-mms22l,grna-plug以及cas9的表达质粒通过lipofectamine3000转染进入前列腺癌细胞系du145中。
在本发明的一个优选实施方式中,我们将grna-mir-155,grna-plug以及cas9的表达质粒通过lipofectamine3000转染进入人胚肾上皮细胞hek293细胞中。
所述的步骤五:加药筛选“塞子”整合入细胞基因组的细胞。具体为:
根据设计的含终止子的供体dna元件(“塞子”)的抗性情况,加药筛选“塞子”整合入细胞基因组的细胞。本发明的方法建议在塞子里加入抗性基因元件(比如neomycin抗性基因,嘌呤霉素抗性基因,潮霉素抗性基因,杀稻瘟菌素抗性基因等),根据实际需要设计构建,有抗性基因可以筛选插入“塞子”的细胞,增加后续单克隆挑选的效率,根据实际需要,也可以不加抗性基因,但效率会降低。除此以外,在该载体上,还可以加入荧光基因等其他报告基因,增加操作者对敲除过程的把握度。
在本发明的一个优选实施方式中,我们导入的“塞子”含有neomycin抗性,且表达egfp。在各个元件导入细胞72小时后,在细胞中加入g418筛选,3周后对照细胞全部死亡,而导入完全基因编辑元件的细胞较好存活,药物筛选成功。
在本发明的一个优选实施方式中,我们导入的“塞子”含有hygromycinb抗性。在各个元件导入细胞72小时后,在细胞中加入hygromycinb筛选,1周后对照细胞全部死亡,而导入完全基因编辑元件的细胞较好存活,药物筛选成功。
所述的步骤六:鉴定“塞子”整合入目的基因设计位置。具体为:
上述筛选成功的细胞分两部分,一部分继续培养,另部分用于鉴定。在基因组切点位置上游(p-gene)和敲入“塞子”的塞子上(p-plug)分别设计引物扩增成功敲入的dna,使得只有外源终止子位于目的基因tss下游的dna可以被扩增出来,用于鉴定是否有细胞的基因组上相应位置成功敲入“塞子”(图1)。
在本发明的一个优选实施方式中,我们拟在du145细胞中敲除ttty15。g418筛选成功的du145细胞经胰酶消化,一半细胞继续培养,另一半细胞用于基因组dna抽提,具体为利用基因组dna抽提试剂盒(qiagen公司),按照说明书步骤抽提,并用针对该优选实施方式的引物:
p-gene-ttty15:tgccgttggtgattcaaggg(seqidno.5)
p-plug:gttgatggcaggggtacgaa(seqidno.6)
通过pcr和测序的方法检测到有细胞的基因组上ttty15转录起始点后成功敲入终止子tk_pa_terminator(图2)。
在本发明的一个优选实施方式中,我们拟在du145细胞中敲除mms22l。g418筛选成功的du145细胞经胰酶消化,一半细胞继续培养,另一半细胞用于基因组dna抽提,具体为利用基因组dna抽提试剂盒(qiagen公司),按照说明书步骤抽提,并用针对该优选实施方式的引物:
p-gene-mms22l:gttctgctgcatcgacgttc(seqidno.7)
p-plug-m:cccgtgagtcaaaccgctat(seqidno.8)
通过pcr和测序的方法检测到有细胞的基因组上mms22l转录起始点后成功敲入终止子tk_pa_terminator(图3)。
在本发明的一个优选实施方式中,我们拟在hek293细胞中敲除mir-155。hygromycinb筛选成功的hek293细胞经胰酶消化,一半细胞继续培养,另一半细胞用于基因组dna抽提,具体为利用基因组dna抽提试剂盒(qiagen公司),按照说明书步骤抽提,并用针对该优选实施方式的引物:
p-gene-mir-155:ccactgggtagtgctttccc(seqidno.9)
p-plug-155:cgggttccttccggtattgt(seqidno.10)
通过pcr和测序的方法检测到有细胞的基因组上mir-155转录起始点后成功敲入终止子tk_pa_terminator(图4)。
所述的步骤七:克隆化成功敲除目的基因的细胞系并鉴定。具体为:
上述六步已经得到部分细胞目的基因被敲除的细胞混合物了,根据需要还可进行单细胞克隆的挑选,并进一步鉴定目的基因被敲除,得到目的基因敲除的细胞株。
pcr鉴定存在成功插入“塞子”的细胞后,说明还在培养的另一部分细胞中也很可能存在携带成功插入“塞子”的细胞,从概率上说,有部分是纯合敲除的细胞(如果是二倍体基因,两条基因都被敲除)。因此,将培养的另一半细胞消化为单细胞悬液,有限稀释法将80个细胞种于96孔板中,镜检标出每孔只有一个细胞的培养孔,继续培养,等细胞长满,消化单克隆细胞,一半细胞每孔细胞对应传至96孔板,另一半细胞标记好在培养孔中所在位置,提基因组dna,用p-gene和p-plug做pcr鉴定。有条带得到说明细胞中存在成功插入“塞子”的基因组。同时设计检测野生型目的基因的引物(p-gene-wt-1,p-gene-wt-2),如果也有条带说明细胞中不是所有目的基因都被敲除(杂合敲除),或者所挑细胞不是单细胞。若p-gene-wt-1,p-gene-wt-2不能p出条带(当然未进行基因编辑操作的对照细胞可以p出),则说明只存在成功插入“塞子”的基因组,不存在野生型目的基因。提示该细胞克隆为纯合敲除单细胞系。进一步通过western-blot、qrt-pcr,southern-blot(dna水平),northern-blot(非编码rna)等方式鉴定是否彻底敲除目的基因。我们发现本发明的方法效率较高,也可跳过单克隆dna水平的鉴定,直接对挑出的克隆进行qrt-pcr水平的鉴定(如实施例2和3)。
在本发明的一个针对ttty15的优选实施方式中,我们在挑出的10个克隆中明确了4个克隆在ttty15下游成功插入含有终止子的“塞子”。选取的两个克隆均不能被野生型ttty15检测引物(p-gene--ttty15-wt-1和p-gene--ttty15-wt-2)扩增出来,说明得到两个可能纯合敲除ttty15的单细胞系。通过qpcr的方式明确目的基因被敲除,图2。
在本发明的一个针对mms22l的优选实施方式中,我们在挑出10个克隆并抽提rna,用qrt-pcr方法鉴定2个mms22l被敲除的细胞株,进一步通过wb明确细胞株中mms22l被敲除,图3。
在本发明的一个针对mir-155的优选实施方式中,我们在挑出10个克隆并抽提rna,用qrt-pcr方法鉴定2个mir-155被敲除的细胞株,图4。
本发明优点在于:
1、本发明提供了一种利用基因编辑技术,敲入终止子实现可转录元件(包括蛋白编码基因和非编码rna等)高效敲除的方法。具体的,通过在拟敲除基因的转录起始位点(tss)后通过基因编辑技术造成dna断裂,同时细胞中导入含有转录终止序列(终止子)的dna供体,该供体不含目的基因同源序列,后者也通过基因编辑技术在细胞类线性化,使得终止子通过非同源修复途径被敲入目的基因tss之后,使得目的基因的内源性转录提前终止,实现目的基因的敲除。
2、本发明方法应用范围广,所有可被转录的元件都可以通过该方法敲除。
3、供体dna具有通用性,效率高,且可包含抗性基因,效率高,有一定的实用价值。
附图说明
图1为利用敲入终止子用于敲除目的基因的策略示意图。
目的基因是可以转录的元件,可以是蛋白编码基因,可以是长链非编码rna母基因,可以是mirna的母基因,以及其他可以被转录的元件。终止子可根据不同的细胞类型和敲除基因类型进行选择,比如真核生物中针对rna聚合酶ii的转录终止可以选择svc0poly(a)signal,beta-globinpoly(a)signal,hsvtkpoly(a)signal,bghpoly(a)signal,tk_pa_terminator等等,或者几个终止子联合运用,针对rna聚合酶iii可以选择连续4个以上的t。比如原核细胞中选择依赖或者不依赖于蛋白质辅因子(如:ρ因子)的转录终止子。抗性基因(比如neomycin抗性基因,嘌呤霉素抗性基因,潮霉素抗性基因,杀稻瘟菌素抗性基因等)。
图2为敲除长链非编码rnattty15的实验结果。
a:g418筛选成功后的细胞挑克隆,抽提dna,利用p-gene-ttty15和p-plug引物行pcr反应,发现10个克隆中有4个克隆在ttty15的转录起始点下游敲入了包含终止子的载体。
b:挑出两个克隆,抽提的dna均不能被p-gene-wt-ttty15-1,p-gene-wt-ttty15-2扩增出来,说明得到两个纯合敲除ttty15的单细胞系。
c:利用p-gene-ttty15和p-plug引物行pcr反应的产物经过测序证明在ttty15的转录起始点下游敲入了包含终止子的载体。
d:通过qpcr的方式明确ttty15被敲除。
图3为敲除蛋白编码基因mms22l的实验结果。
a:g418筛选成功后的混合细胞抽提基因组dna,经由引物p-gene-mms22l和p-plug-m进行pcr扩增发现转染“塞子”等质粒后有目标条带,提示“塞子”被插入mms22l转录起始点后。
我们在挑出10个克隆并抽提rna,用qrt-pcr方法鉴定2个mms22l被敲除的细胞株。
western-blot证实挑选出一个单细胞克隆(clone4)中mms22l被敲除。
图4为敲除micrornamir-155的实验结果。
a:hygromycinb筛选成功后的混合细胞抽提基因组dna,经由引物p-gene-mir-155和plug-155进行pcr扩增发现转染“塞子”等质粒后出现预期大小条带,提示“塞子”被插入mir-155转录起始点后。
b:我们在挑出10个克隆并抽提rna,用利用颈环引物反转录后qrt-pcr方法鉴定到clone1和clone4这2个细胞株的mir155被敲除。
具体实施方式
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。
下列实施例中未注明具体条件的实验方法,通常按照常规条件如sambrook等人,分子克隆:实验室指南(newyork:coldspringharborlaboratorypress,1989)中所述的条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。除非另行定义,文中所使用的所有专业与科学用语与本领域熟练人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法及材料皆可应用于本发明中。文中所述的较佳实施方法与材料仅作示范之用。
实施例1:crispr-cas9技术敲入终止子实现长链非编码rnattty15的敲除
1)利用分子克隆技术构建一个包含转录终止序列(终止子)tk_pa_terminator和含有neomycin抗性基因,并表达egfp的载体。
其中tk_pa_terminator的具体序列如seqidno.1所示。
2)选择了哺乳动物中的人类的基因组作为设计干预目标,ttty15作为拟敲除的非编码rna,设计识别grna:grna-ttty15,其识别位点序列如下,tcaggcaatccagccgcccggg(seqidno.2),其中ggg为pam序列。
3)分别构建grna-ttty15和grna-plug表达载体,相应的表达元件被构建到慢病毒骨架载体plko-bsd中(这两个质粒可以被包装成慢病毒)。cas9表达载体为商品化的质粒cas9表达载体购自addgene(plasmidid号:41815)。
4)将grna-ttty15,grna-plug以及cas9的表达质粒通过lipofectamine3000转染进入前列腺癌细胞系du145中。
5)在各个元件导入du145细胞72小时后,在细胞中加入g418筛选,3周后对照细胞全部死亡,而导入完全基因编辑元件的细胞较好存活,药物筛选成功。
6)g418筛选成功的du145细胞经胰酶消化,一半细胞继续培养,另一半细胞用于基因组dna抽提,具体为利用基因组dna抽提试剂盒(qiagen公司),按照说明书步骤抽提,并用针对本实施例的引物:
p-gene-ttty15:tgccgttggtgattcaaggg(seqidno.5),该条引物结合ttty15切点上游,位于基因组上。
p-plug:gttgatggcaggggtacgaa(seqidno.6),该条引物位于“塞子”上。
通过pcr和测序的方法检测是否有目标条带,如果有证明有细胞的基因组上ttty15转录起始点后成功敲入终止子tk_pa_terminator(图2)。
pcr反应体系加样:
经过预实验后,最终确定以下条件:
pcr反应结束后,进行琼脂糖凝胶电泳,发现对照细胞的dna不能扩增出任何条带,而实验组扩增出预期大小条带。
7)克隆化成功敲除目的基因的细胞系并鉴定。
上述六步已经得到部分细胞目的基因被敲除的细胞混合物了,根据需要还可进行单细胞克隆的挑选,并进一步鉴定目的基因被敲除,得到目的基因敲除的细胞株。
pcr鉴定存在成功插入“塞子”的细胞后,说明还在培养的另一部分细胞中也很可能存在携带成功插入“塞子”的细胞,从概率上说,有部分是纯合敲除的细胞(如果是二倍体基因,两条基因都被敲除)。因此,将培养的另一半细胞消化为单细胞悬液,有限稀释法将80个细胞种于96孔板中,镜检标出每孔只有一个细胞的培养孔,继续培养,等细胞长满,消化单克隆细胞,一半细胞每孔细胞对应传至96孔板,另一半细胞标记好在培养孔中所在位置,提基因组dna。我们在挑出10个克隆并抽提dna,用p-gene-ttty15和p-plug做pcr鉴定,发现4个克隆在ttty15下游成功插入含有终止子的“塞子”(图2a,c)。
同时设计检测野生型目的基因的引物:
p-gene--ttty15-wt-1:aacggtgattgcgtgaatgtt(seqidno.11)
p-gene--ttty15-wt-2:cctgagcctcctacattatctcac(seqidno.12)
发现在挑出的10个克隆中明确了4个克隆在ttty15下游成功插入含有终止子的“塞子”。选取的两个克隆均不能被p-gene-wt-1,p-gene-wt-2扩增出来,说明得到两个可能纯合敲除ttty15的单细胞系(图2b)。通过qpcr的方式明确目的基因被敲除(图2d)。
实施例2:crispr-cas9技术敲入终止子实现蛋白编码基因mms22l的敲除
1)利用分子克隆技术构建一个包含转录终止序列(终止子)tk_pa_terminator和含有neomycin抗性基因,并表达egfp的载体。
2)选择了哺乳动物中的人类的基因组作为设计干预目标,mms22l作为拟敲除的蛋白编码基因,设计识别grna:grna-mms22l,其识别位点序列如下,gcttaagggctccgctgcagagg(seqidno.3),其中agg为pam序列。
3)分别构建grna-mms22l和grna-plug的表达载体,相应的表达元件被构建到慢病毒骨架载体plko-bsd中(这两个质粒可以被包装成慢病毒)。cas9表达载体为商品化的质粒cas9表达载体购自addgene(plasmidid号:41815)。
4)将grna-mms22l,grna-plug以及cas9的表达质粒通过lipofectamine3000转染进入前列腺癌细胞系du145中。
5)在各个元件导入du145细胞72小时后,在细胞中加入g418筛选,3周后对照细胞全部死亡,而导入完全基因编辑元件的细胞较好存活,药物筛选成功。
6)g418筛选成功的du145细胞经胰酶消化,一半细胞继续培养,另一半细胞用于基因组dna抽提,具体为利用基因组dna抽提试剂盒(qiagen公司),按照说明书步骤抽提,进行pcr反应,用针对本实施例的引物:
p-gene-mms22l:gttctgctgcatcgacgttc(seqidno.7),该条引物结合mms22l切点上游,位于基因组上。
p-plug-m:cccgtgagtcaaaccgctat(seqidno.8),该条引物位于“塞子”上。
pcr反应结束后,进行琼脂糖凝胶电泳,其结果如图3所示,可见对照细胞的dna不能扩增出任何条带,而实验组扩增出预期大小条带。证明有细胞的基因组上mms22l转录起始点后成功敲入终止子tk_pa_terminator。
7)克隆化成功敲除目的基因的细胞系并鉴定。
上述六步已经得到部分细胞目的基因被敲除的细胞混合物了,根据需要还可进行单细胞克隆的挑选,并进一步鉴定目的基因被敲除,得到目的基因敲除的细胞株。
pcr鉴定存在成功插入“塞子”的细胞后,说明还在培养的另一部分细胞中也很可能存在携带成功插入“塞子”的细胞,从概率上说,有部分是纯合敲除的细胞(如果是二倍体基因,两条基因都被敲除)。因此,将培养的另一半细胞消化为单细胞悬液,有限稀释法将80个细胞种于96孔板中,镜检标出每孔只有一个细胞的培养孔,继续培养,等细胞长满,消化单克隆细胞,一半细胞标记好在培养孔中所在位置,另一半细胞传至24孔板,提基因组rna。我们在挑出10个克隆并抽提rna,用qrt-pcr方法鉴定2个mms22l被敲除的细胞株,见图3b。
进一步通过wb明确细胞株中mms22l被敲除,见图3c。
实施例3:crispr-cas9技术敲入终止子实现micrornamir-155的敲除
1)利用分子克隆技术构建一个包含转录终止序列(终止子)tk_pa_terminator(侧翼含tttt序列)和含有hygromycinb抗性基因的载体。
2)选择了哺乳动物中的人类的基因组作为设计干预目标,mir-155作为拟敲除的microrna,设计识别grna:grna-mir-155,其识别位点序列如下,gttaatgctaatcgtgatagggg(seqidno.4),其中ggg为pam序列。
3)分别构建grna-mir-155和grna-plug的表达载体,相应的表达元件被构建到慢病毒骨架载体plko-bsd中(这两个质粒可以被包装成慢病毒)。cas9表达载体为商品化的质粒cas9表达载体购自addgene(plasmidid号:41815)。
4)将grna-mir-155,grna-plug以及cas9的表达质粒通过lipofectamine3000转染进入人胚肾上皮细胞hek293细胞中。
5)在各个元件导入hek293细胞72小时后,在细胞中加入hygromycinb筛选,1周后对照细胞全部死亡,而导入完全基因编辑元件的细胞较好存活,药物筛选成功。
6)hygromycinb筛选成功的hek293细胞经胰酶消化,一半细胞继续培养,另一半细胞用于基因组dna抽提,具体为利用基因组dna抽提试剂盒(qiagen公司),按照说明书步骤抽提,并用针对本实施例的引物:
p-gene-mir-155:ccactgggtagtgctttccc(seqidno.9),该条引物结合mir-155切点上游,位于基因组上。
p-plug-155:cgggttccttccggtattgt(seqidno.10),该条引物位于“塞子”上。
通过pcr和测序的方法检测发现有目标条带证明有细胞的基因组上mir-155转录起始点后成功敲入终止子tk_pa_terminator。
pcr反应结束后,进行琼脂糖凝胶电泳,其结果如图4a所示,可见对照细胞的dna不能扩增出任何条带,而实验组扩增出预期大小条带。
7)克隆化成功敲除目的基因的细胞系并鉴定。
上述六步已经得到部分细胞目的基因被敲除的细胞混合物了,根据需要还可进行单细胞克隆的挑选,并进一步鉴定目的基因被敲除,得到目的基因敲除的细胞株。
pcr鉴定存在成功插入“塞子”的细胞后,说明还在培养的另一部分细胞中也很可能存在携带成功插入“塞子”的细胞,从概率上说,有部分是纯合敲除的细胞(如果是二倍体基因,两条基因都被敲除)。因此,将培养的另一半细胞消化为单细胞悬液,有限稀释法将80个细胞种于96孔板中,镜检标出每孔只有一个细胞的培养孔,继续培养,等细胞长满,消化单克隆细胞,一半细胞标记好在培养孔中所在位置,另一半细胞传至24孔板,提基因组rna。我们在挑出10个克隆并抽提rna,用qrt-pcr方法鉴定2个mir-155被敲除的细胞株,见图4b。
以上已对本发明创造的较佳实施例进行了具体说明,但本发明创造并不限于所述实施例,熟悉本领域的技术人员在不违背本发明创造精神的前提下还可做出种种的等同的变型或替换,这些等同的变型或替换均包含在本申请权利要求所限定的范围内。
序列表
<110>上海长海医院
<120>一种利用基因编辑技术敲入终止子实现可转录元件敲除的方法
<130>/
<160>12
<170>siposequencelisting1.0
<210>1
<211>493
<212>dna
<213>人工序列(artificial)
<400>1
tgaaaggttgggcttcggaatcgttttccgggacgccggctggatgatcctccagcgcgg60
ggatctcatgctggagttcttcgcccaccctagggggaggctaactgaaacacggaagga120
gacaataccggaaggaacccgcgctatgacggcaataaaaagacagaataaaacgcacgg180
tgttgggtcgtttgttcataaacgcggggttcggtcccagggctggcactctgtcgatac240
cccaccgagaccccattggggccaatacgcccgcgtttcttccttttccccaccccaccc300
cccaagttcgggtgaaggcccagggctcgcagccaacgtcggggcggcaggccctgccat360
agcctcaggttactcatatatactttagattgatttaaaacttcatttttaatttaaaag420
gatctaggtgaagatcctttttgataatctcatgaccaaaatcccttaacgtgagttttc480
gttccactgagcg493
<210>2
<211>22
<212>dna
<213>人工序列(artificial)
<400>2
tcaggcaatccagccgcccggg22
<210>3
<211>23
<212>dna
<213>人工序列(artificial)
<400>3
gcttaagggctccgctgcagagg23
<210>4
<211>23
<212>dna
<213>人工序列(artificial)
<400>4
gttaatgctaatcgtgatagggg23
<210>5
<211>20
<212>dna
<213>人工序列(artificial)
<400>5
tgccgttggtgattcaaggg20
<210>6
<211>20
<212>dna
<213>人工序列(artificial)
<400>6
gttgatggcaggggtacgaa20
<210>7
<211>20
<212>dna
<213>人工序列(artificial)
<400>7
gttctgctgcatcgacgttc20
<210>8
<211>20
<212>dna
<213>人工序列(artificial)
<400>8
cccgtgagtcaaaccgctat20
<210>9
<211>20
<212>dna
<213>人工序列(artificial)
<400>9
ccactgggtagtgctttccc20
<210>10
<211>20
<212>dna
<213>人工序列(artificial)
<400>10
cgggttccttccggtattgt20
<210>11
<211>21
<212>dna
<213>人工序列(artificial)
<400>11
aacggtgattgcgtgaatgtt21
<210>12
<211>24
<212>dna
<213>人工序列(artificial)
<400>12
cctgagcctcctacattatctcac24
- 还没有人留言评论。精彩留言会获得点赞!