一种4-乙基吡啶的制备方法与流程
本发明涉及有机合成技术领域,尤其涉及一种4-乙基吡啶的制备方法。
背景技术:
乙基吡啶是许多药物合成的起始原料,特别是现在很多杀虫剂药物合成的中间体,但是还没有工业化生产4-乙烯基吡啶的报道。
目前,实验室制备4-乙基吡啶的常用方法有:
(1)在铁粉或锌粉存在下,用醋酐使吡啶烃化制得4-乙基吡啶。该方法在反应过程中,需要消耗大量的醋酸,同时分离产物时需要消耗大量的碳酸钾,不仅后处理比较困难,而且得到的4-乙基吡啶的收率也较低,约为35%~45.5%。
(2)将4-乙烯基吡啶在贵金属催化剂下进行高压加氢,制备4-乙基吡啶。该方法虽然成本低,但是4-乙烯基吡啶很容易聚合,该方法很难实行工业化,属于危险工艺。
技术实现要素:
为了解决现有技术中的问题,本发明提供了一种可工业化,且收率高的4-乙基吡啶的制备方法,本发明提供的方法可应用于工业生产中,且收率较高。
本发明提供了一种4-乙基吡啶的制备方法,包括以下步骤:
(1)将4-吡啶甲酸乙酯和乙醇钠混合后,加热至90~110℃,然后滴加乙酸乙酯,进行克莱森酯缩合反应,得到3-氧代3-(4-吡啶基)丙酸乙酯;
(2)将所述步骤(1)得到的3-氧代3-(4-吡啶基)丙酸乙酯、二甲基亚砜和水混合后,进行加热处理,得到4-乙酰基吡啶;
(3)将乙二醇和氢氧化钾混合后冷却,加入水合肼,升温至60~80℃,然后与所述步骤(2)得到的4-乙酰基吡啶混合,进行还原反应,得到4-乙基吡啶。
优选的,所述步骤(1)中4-吡啶甲酸乙酯和乙酸乙酯的质量比为75~80:45~55。
优选的,所述步骤(1)中4-吡啶甲酸乙酯和乙醇钠的质量比为75~80:20~25。
优选的,所述步骤(1)中克莱森酯缩合反应的温度为90~110℃,时间为4~4.5h。
优选的,所述步骤(2)中加热处理的温度为150~160℃,时间为1~2h。
优选的,所述步骤(2)中3-氧代3-(4-吡啶基)丙酸乙酯、二甲基亚砜和水的用量比为2.5~3g:0.5~1.5ml:1.5~2.5ml。
优选的,所述步骤(3)中乙二醇和氢氧化钾的用量比为2~3ml:1g;所述水合肼和4-乙酰基吡啶的摩尔比为1~1.1:1。
优选的,所述步骤(3)中还原反应在梯度升温程序下进行,所述梯度升温程序为:
85~94℃反应1~2h;
95~104℃反应1~2h;
105~114℃反应1~2h
115~124℃反应1~2h。
优选的,所述步骤(1)中4-吡啶甲酸乙酯的制备方法包括以下步骤:
将异烟酸、无水乙醇、活化后的活性炭和甲苯混合,进行微波反应,得到4-吡啶甲酸乙酯;所述微波反应的功率为180~220w,时间为8~12min;
所述活化后的粉末活性炭的制备方法为:采用对甲基苯磺酸水溶液对活性炭进行浸泡处理后,依次进行过滤和干燥得到。
优选的,所述异烟酸和无水乙醇的摩尔比为1:0.5~1.0。
本发明提供了一种4-乙基吡啶的制备方法,包括以下步骤:将4-吡啶甲酸乙酯和乙醇钠混合后,加热至90~110℃,然后滴加乙酸乙酯,进行克莱森酯缩合反应,得到3-氧代3-(4-吡啶基)丙酸乙酯;将3-氧代3-(4-吡啶基)丙酸乙酯、二甲基亚砜和水混合后,进行加热处理,得到4-乙酰基吡啶;将乙二醇和氢氧化钾混合后冷却,加入水合肼,升温至60~80℃,然后与4-乙酰基吡啶混合,进行还原反应,得到4-乙基吡啶。本发明提供了一种制备4-乙基吡啶的新方法,本发明提供的方法不仅可以应用在工业上,而且本发明提供的方法制备得到的4-乙酰基吡啶收率和纯度较高,收率达88~89%,纯度达为99~99.9%。
附图说明
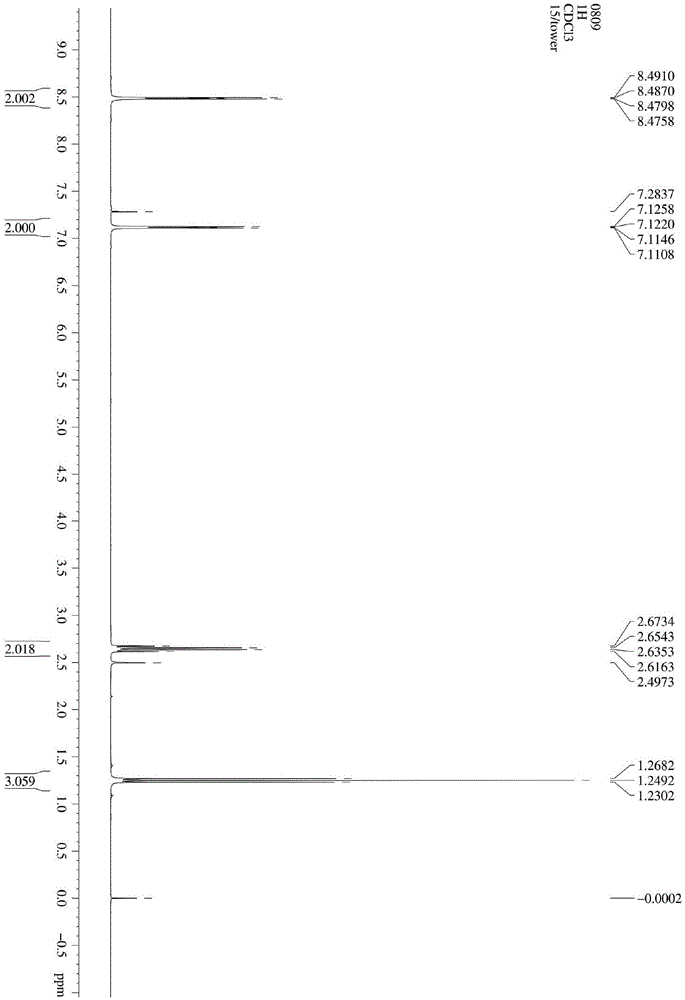
图1为本发明实施例1制备得到的4-乙基吡啶的氢核磁谱图;
图2为本发明实施例1制备得到的4-乙基吡啶的气相色谱图。
具体实施方式
本发明提供了一种4-乙基吡啶的制备方法,包括以下步骤:
(1)将4-吡啶甲酸乙酯和乙醇钠混合后,加热至90~110℃,然后滴加乙酸乙酯,进行克莱森酯缩合反应,得到3-氧代3-(4-吡啶基)丙酸乙酯;
(2)将所述步骤(1)得到的3-氧代3-(4-吡啶基)丙酸乙酯、二甲基亚砜和水混合后,进行加热处理,得到4-乙酰基吡啶;
(3)将乙二醇和氢氧化钾混合后冷却,加入水合肼,升温至60~80℃,然后与所述步骤(2)得到的4-乙酰基吡啶混合,进行还原反应,得到4-乙基吡啶。
本发明将4-吡啶甲酸乙酯和乙醇钠混合后,加热至90~110℃,然后滴加乙酸乙酯,进行克莱森酯缩合反应,得到3-氧代3-(4-吡啶基)丙酸乙酯。
在本发明中,所述4-吡啶甲酸乙酯的制备方法优选包括以下步骤:将异烟酸、无水乙醇、活化后的活性炭和甲苯混合,进行微波反应,得到4-吡啶甲酸乙酯。
在本发明中,所述活化后的活性炭的制备方法优选为:采用对甲基苯磺酸水溶液对活性炭进行浸泡处理后,依次进行过滤和干燥,得到活化后的活性炭。在本发明中,所述对甲基苯磺酸水溶液的质量浓度优选为20%~30%,进一步优选为25%,所述浸泡处理的时间优选为28~32h,进一步优选为30h。在本发明中,所述活性炭优选为粉末活性炭,本发明对粉末活性炭的粒径没有要求。本发明对过滤和干燥的具体实施方式没有特殊要求,采用本领域技术人员所熟知的即可。
本发明将异烟酸、无水乙醇、活化后的活性炭和甲苯混合,所述异烟酸和无水乙醇的摩尔比优选为1:0.5~1.0,进一步优选为1:0.8~0.85;所述异烟酸和催化剂的用量比优选为0.8mol:1.8~2.2g。
混合后,本发明将所述混合物进行微波反应,所述微波反应的功率优选为180~220w,进一步优选为200w,时间优选为8~12min,进一步优选为10min。微波反应完成后,本发明优选将所述微波反应产物的ph值调节至6.5~7.5,更优选为7.0,然后静置分层,将上层有机层进行蒸馏,得到4-吡啶甲酸乙酯。本发明优选采用饱和na2co3水溶液调节微波反应产物的ph值,本发明优选将静置分层得到的水相用氯仿萃取,萃取液与上层有机层合并后,进行蒸馏。在本发明中,所述蒸馏优选包括依次进行的常压蒸馏和减压蒸馏,本发明优选通过常压蒸馏回收氯仿,然后通过减压蒸馏,得到4-吡啶甲酸乙酯。
本发明制备4-吡啶甲酸乙酯的反应式如式i所示:
在本发明中,所述4-吡啶甲酸乙酯的收率为97~98%,纯度为96~97%,所述纯度优选通过气相色谱法测试得到。
得到4-吡啶甲酸乙酯后,本发明将4-吡啶甲酸乙酯和乙醇钠混合后,加热至90~110℃,优选为95~105℃,更优选为100℃;然后滴加乙酸乙酯,进行克莱森酯缩合反应,得到反应产物。在本发明中,所述4-吡啶甲酸乙酯和乙醇钠的质量比优选为75~80:20~25,进一步优选为76~78:20~23,更优选为76:20.4;所述4-吡啶甲酸乙酯和乙酸乙酯的质量比优选为75~80:45~55,进一步优选为76~78:48~52,更优选为76:50。在本发明中,所述克莱森酯缩合反应的温度优选为90~110℃,进一步优选为95~105℃,更优选为100℃,时间优选为4~4.5h,所述克莱森酯缩合反应的时间优选从滴加完乙酸乙酯开始计算。本发明通过所述克莱森酯缩合反应得到的反应产物为粘稠状物质。
得到反应产物后,本发明优选调节所述反应产物的ph值为6.5~7.5,在本发明中,所述反应产物的ph值更优选调节至6.8~7.2,进一步优选调节至7.0。本发明优选将所述反应产物的ph值调节到上述范围内,是因为如果ph值过低,酸性过大会导致下一步反应无法进行。本发明优选采用稀盐酸调节反应产物的ph值,本发明对稀盐酸的浓度没有特殊要求,只要能够将反应产物的ph值调节到上述范围即可。
调节完ph值后,本发明优选对调节ph值后的反应产物依次进行过滤、滤饼洗涤和抽滤处理,得到3-氧代3-(4-吡啶基)丙酸乙酯。本发明对过滤、洗涤和抽滤的具体实施方式没有特别限制,采用本领域技术人员所熟知的方式即可。在本发明中,所述洗涤用洗涤剂优选为石油醚。
本发明以4-吡啶甲酸乙酯为原料制备得到3-氧代3-(4-吡啶基)丙酸乙酯的反应式如式ii所示:
在本发明中,所述3-氧代3-(4-吡啶基)丙酸乙酯的收率为93%~95%,纯度为95~96%,所述纯度优选通过气相色谱法测试得到。
得到3-氧代3-(4-吡啶基)丙酸乙酯后,本发明将3-氧代3-(4-吡啶基)丙酸乙酯、二甲基亚砜和水混合,然后进行加热处理,得到4-乙酰基吡啶。
在本发明中,所述3-氧代3-(4-吡啶基)丙酸乙酯、二甲基亚砜和水的用量比优选为2.5~3g:0.5~1.5ml:1.5~2.5ml,进一步优选为2.5~3g:1ml:2ml。在本发明中,所述加热处理的温度优选为150~160℃,进一步优选为152~158℃,更优选为155℃,时间优选为1~2h,进一步优选为1.5~2h。
加热处理完成后,得到加热反应产物。本发明优选向加热反应产物中加入水和过量氯化钠,以形成氯化钠饱和溶液,然后进行乙酸乙酯萃取,直至萃取得到的水相中没有产物4-乙酰基吡啶,将所得乙酸乙酯相用饱和食盐水洗涤后,干燥浓缩,得到4-乙酰基吡啶。在本发明中,所述乙酸乙酯萃取的次数优选为2~3次。
在本发明中,所述3-氧代3-(4-吡啶基)丙酸乙酯制备得到4-乙酰基吡啶的反应式如式iii所示:
在本发明中,所述4-乙酰基吡啶的收率为85~86%,纯度为93~94%,所述纯度优选通过气相色谱法测试得到。
得到4-乙酰基吡啶后,本发明将乙二醇和氢氧化钾混合后冷却,加入水合肼,升温至60~80℃,然后加入制备得到的4-乙酰基吡啶,进行还原反应,得到4-乙基吡啶。
本发明先将乙二醇和氢氧化钾混合,所述乙二醇和氢氧化钾的用量比优选为2~3ml:1g,进一步优选为2.5ml:1g;所述乙二醇和氢氧化钾在混合过程中,释放大量热量,本发明将混合后的乙二醇和氢氧化钾冷却至室温后,加入水合肼。在本发明中,所述水合肼和4-乙酰基吡啶的摩尔比优选为1~1.1:1,进一步优选为1.05:1。在本发明中,所述4-乙酰基吡啶和氢氧化钾的摩尔比优选为1:0.8~1.2,进一步优选为1:1。本发明加入水合肼后,优选升温至65~75℃,进一步优选为70℃。在本发明中,所述还原反应优选在梯度升温程序下进行,所述梯度升温程序优选为:85~94℃反应1~2h;95~104℃反应1~2h;105~114℃反应1~2h;115~124℃反应1~2h。
还原反应完成后,本发明优选调节还原反应产物的ph值至6.5~7.5,优选为7.0。在本发明中,调节还原反应产物ph值的调节剂优选为冰醋酸。调节完还原产物的ph值后,本发明优选向调节ph值后的还原反应产物中加入水和过量氯化钠,以形成氯化钠饱和溶液,然后进行乙酸乙酯萃取,直至萃取得到的水相中没有产物4-乙基吡啶,将所得乙酸乙酯相用饱和食盐水洗涤后,干燥浓缩,减压蒸馏,收集76~80℃的馏出物,得到4-乙基吡啶。在本发明中,所述乙酸乙酯萃取的次数优选为2~3次。本发明对干燥浓缩和减压蒸馏的具体实施方式没有特殊限制,采用本领域技术人员所熟知的即可。
在本发明中,所述4-乙酰基吡啶制备得到4-乙基吡啶的反应式如式iv所示:
在本发明中,所述4-乙基吡啶的收率为88~89%,纯度为99~99.9%,所述纯度优选通过气相色谱测试得到。
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。
实施例1
(1)制备4-吡啶甲酸乙酯
在100ml三口瓶中依次加入0.8mol异烟酸、0.64mol无水乙醇、2.0g活化后的粉末活性炭作催化剂,甲苯作溶剂。微波反应时间10min(功率200w;温度130℃)。用饱和na2co3水溶液中和反应液至ph为7,静置分层,取上层机层;水相用20ml氯仿萃取,合并萃取液与有机层。先常压蒸馏回收氯仿再减压蒸馏得无色透明液体4-吡啶甲酸乙酯,纯度为96.3%(gc),收率为97.2%。
活化后的粉末状活性炭通过将粉末活性炭浸泡在60ml25%对甲基苯磺酸水溶液中30h,抽滤,干燥得到。
(2)制备3-氧代3-(4-吡啶基)丙酸乙酯
4-吡啶甲酸乙酯和乙酸乙酯发生克莱森酯缩合反应,另一三口瓶中配置温度计,机械搅拌,室温加入4-吡啶甲酸乙酯76g,乙醇钠20.4克,油浴加热,反应瓶内温度由25℃缓慢升温到100℃左右,滴加乙酸乙酯50克,滴加完毕后,约4小时反应完毕,体系较粘稠。冷却至室温,稀盐酸调节ph=7后搅拌半小时,过滤,固体用石油醚洗涤;抽滤所得固体为3-氧代3-(4-吡啶基)丙酸乙酯,纯度95.4%(gc),收率93.4%。
(3)制备4-乙酰基吡啶
向反应瓶中加入3-氧代3-(4-吡啶基)丙酸乙酯和二甲基亚砜,其中3-氧代3-(4-吡啶基)丙酸乙酯为2.5g,二甲基亚砜的体积为1ml,然后加入2ml水,油浴加热,155℃反应1.5小时,反应完毕后,加入水稀释,再加过量nacl处理成饱和食盐水,ea(乙酸乙酯)萃取2-3次,至水相无产品,ea相用饱和食盐水洗涤两次后,干燥浓缩,得到纯度为93.6%(gc),收率为85.3%的4-乙酰基吡啶。
(4)制备4-乙基吡啶
向反应瓶投入2.5ml乙二醇和1g氢氧化钾,搅拌溶解,放热明显,澄清后,冷至室温,加入1.17g质量浓度为80%的水合肼,升温至70℃,滴加2.17g4-乙酰吡啶;缓慢升温,分别在90℃,100℃,110℃各反应1小时,在120℃反应2小时,反应结束后,加入冰醋酸调节ph=7,加入乙二醇体积3倍的水稀释后,加过量nacl处理成饱和食盐水,ea萃取2-3次,至水相无产品,ea相用饱和食盐水洗涤两次后,干燥浓缩,减压蒸馏,水泵76~80℃为产品,收率88.6%。对实施例1制备得到的产品进行氢核磁表征,结果如图1所示,由图1可知本发明制备得到的产品为4-乙基吡啶。对实施例1制备得到的产品纯度进行气相色谱(gc)检测,检测纯度为99.2%,结果如图2所示。
实施例2
(1)制备4-吡啶甲酸乙酯
在100ml三口瓶中依次加入0.8mol异烟酸、0.64mol无水乙醇、2.0g活化后的粉末活性炭作催化剂,甲苯作溶剂。微波反应时间10min(功率200w;温度130℃)。用饱和na2co3水溶液中和反应液至ph为7,静置分层,取上层机层;水相用20ml氯仿萃取,合并萃取液与有机层。先常压蒸馏回收氯仿再减压蒸馏得无色透明液体4-吡啶甲酸乙酯,纯度为96.3%(gc),收率为97.2%。
活化后的粉末状活性炭通过将粉末活性炭浸泡在60ml25%对甲基苯磺酸水溶液中30h,抽滤,干燥得到。
(2)制备3-氧代3-(4-吡啶基)丙酸乙酯
4-吡啶甲酸乙酯和乙酸乙酯发生克莱森酯缩合反应,另一三口瓶中配置温度计,机械搅拌,室温加入4-吡啶甲酸乙酯76g,乙醇钠20.4克,油浴加热,反应瓶内温度由25℃缓慢升温到105℃,滴加乙酸乙酯50克,滴加完毕后,约4小时反应完毕,体系较粘稠。冷却至室温,稀盐酸调节ph=6.8后搅拌半小时,过滤,固体用石油醚洗涤;抽滤所得固体为3-氧代3-(4-吡啶基)丙酸乙酯,纯度95.5%(gc),收率93.5%。
(3)制备4-乙酰基吡啶
向反应瓶中加入3-氧代3-(4-吡啶基)丙酸乙酯和二甲基亚砜,其中3-氧代3-(4-吡啶基)丙酸乙酯为2.5g,二甲基亚砜的体积为1ml,然后加入水,水的用量为2ml,油浴加热,150℃反应2小时,反应完毕后,加入水稀释,再加过量nacl处理成饱和食盐水,ea(乙酸乙酯)萃取2-3次,至水相无产品,ea相用饱和食盐水洗涤两次后,干燥浓缩,得到纯度为94%(gc),收率为85.5%的4-乙酰基吡啶。
(4)制备4-乙基吡啶
向反应瓶投入2.5ml乙二醇和1g氢氧化钾,搅拌溶解,放热明显,澄清后,冷至室温,加入1.17g质量浓度为80%的水合肼,升温至75℃,滴加2.17g4-乙酰吡啶;缓慢升温,分别在90℃,100℃,110℃各反应1.5小时,在120℃反应2小时,反应结束后,加入冰醋酸调节ph=6.8,加入乙二醇体积3倍的水稀释后,加过量nacl处理成饱和食盐水,ea萃取2-3次,至水相无产品,ea相用饱和食盐水洗涤两次后,干燥浓缩,减压蒸馏,水泵76~80℃为产品,gc检测纯度为99.5%,收率89%。
实施例3
(1)制备4-吡啶甲酸乙酯
在100l反应釜中依次加入98.4kg异烟酸、29.4kg无水乙醇、2kg活化后的粉末活性炭作催化剂,甲苯作溶剂。微波反应时间10min(功率200w;温度130℃)。用饱和na2co3水溶液中和反应液至ph为7,静置分层,取上层机层;水相用氯仿萃取,合并萃取液与有机层。先常压蒸馏回收氯仿再减压蒸馏得无色透明液体4-吡啶甲酸乙酯,纯度为96.3%(gc),收率为97.2%。
活化后的粉末状活性炭通过将粉末活性炭浸泡在60l25%对甲基苯磺酸水溶液中30h,抽滤,干燥得到。
(2)制备3-氧代3-(4-吡啶基)丙酸乙酯
4-吡啶甲酸乙酯和乙酸乙酯发生克莱森酯缩合反应,另一三口瓶中配置温度计,机械搅拌,室温加入4-吡啶甲酸乙酯76kg,乙醇钠20.4kg,油浴加热,反应瓶内温度由25℃缓慢升温到100℃左右,滴加乙酸乙酯50kg,滴加完毕后,约4小时反应完毕,体系较粘稠。冷却至室温,稀盐酸调节ph=7后搅拌半小时,过滤,固体用石油醚洗涤;抽滤所得固体为3-氧代3-(4-吡啶基)丙酸乙酯,纯度95.4%(gc),收率93.4%。
(3)制备4-乙酰基吡啶
向反应瓶中加入2.5kg3-氧代3-(4-吡啶基)丙酸乙酯和1l二甲基亚砜,然后加入2l水,油浴加热,155℃反应1.5小时,反应完毕后,加入水稀释,再加过量nacl处理成饱和食盐水,ea(乙酸乙酯)萃取2-3次,至水相无产品,ea相用饱和食盐水洗涤两次后,干燥浓缩,得到纯度为93.6%(gc),收率为85.3%的4-乙酰基吡啶。
(4)制备4-乙基吡啶
向反应瓶投入2.5l乙二醇和1kg氢氧化钾,搅拌溶解,放热明显,澄清后,冷至室温,加入1.17kg质量浓度为80%的水合肼,升温至70℃,滴加2.17kg4-乙酰吡啶;缓慢升温,分别在90℃,100℃,110℃各反应1小时,在120℃反应2小时,反应结束后,加入冰醋酸调节ph=7,加入乙二醇体积3倍的水稀释后,加过量nacl处理成饱和食盐水,ea萃取2-3次,至水相无产品,ea相用饱和食盐水洗涤两次后,干燥浓缩,减压蒸馏,水泵76~80℃为产品,gc检测纯度为99.2%,收率88.6%。
对实施例2~3制备得到的产品进行氢核磁表征,结果与图1相同,在此不再赘述,由此可知,本发明实施例2~3制备得到的产品为4-乙基吡啶。
对比例1
按照实施例1的方法进行试验,区别在于,在制备3-氧代3-(4-吡啶基)丙酸乙酯时,采用稀盐酸调节ph=5,最终得到的是液体,无法得到3-氧代3-(4-吡啶基)丙酸乙酯。
综上,本发明提供的方法收率和纯度较高,且可以应用于工业生产。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!