用于检测由融合基因和/或外显子跳跃产生的转录产物的探针以及方法与流程
本发明涉及用于判定基因组上的融合基因的转录产物的存在或其表达量的探针、用于判定由外显子跳跃产生的转录产物的存在或其表达量的探针、包含该探针的试剂盒、使用该探针来判定基因组上的融合基因的转录产物的存在或其表达量的方法、以及判定由外显子跳跃产生的转录产物的存在或其表达量的方法等。
背景技术:
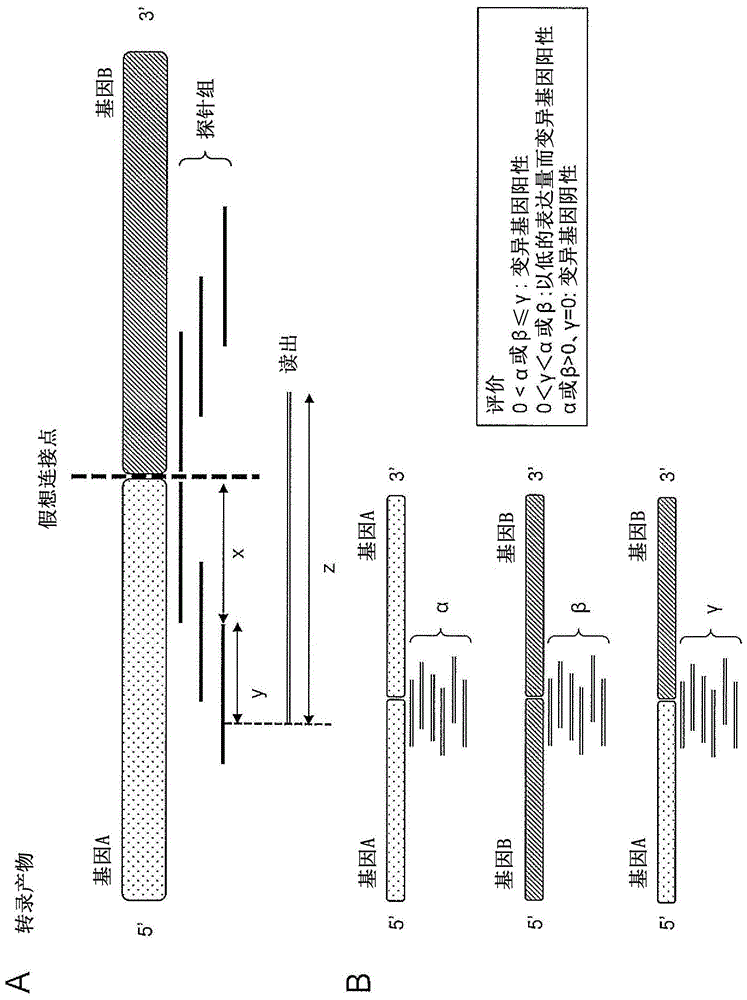
:融合基因作为体细胞癌症变异的一个原因而已知,目前为止,针对起因于融合基因的癌症研发出了几种治疗法。例如,可列举针对具有慢性骨髓白血病中的bcr-abl1融合基因(非专利文献1)、非小细胞肺癌中的eml4-alk融合基因(非专利文献2)等的癌症变异的患者使用酪氨酸激酶抑制剂的第一选择疗法。由此,改善了起因于融合基因的癌症的治疗成绩。通过近年来的测序技术的进步,能够进行癌症基因组以及转录组中的染色体重排的全面检测,发现了ret、ros1、ntrk1、nrg1或fgrf1/2/3基因等的融合基因(非专利文献3~8),也使这些融合基因应用于癌症的诊断。此外,表明了近年来除了融合基因之外,met14外显子跳跃等的外显子跳跃也可能成为癌症的原因。然而,由于这些融合基因以及外显子跳跃的发生为比较低的频率,其种类也多样,因此难以同时检测成为靶向基因的多个融合基因。此外,由于fish、免疫组织化学以及逆转录pcr等的以往方法的诊断需要专业技术,因此为了临床应用而迫切期望能够简便地检测多个靶向基因的方法。基于通过扩增子pcr或杂交捕获进行的gdna的靶向基因富集的癌症相关基因的靶向测序,是融合基因等的变异的检测所使用的方法的一例。然而,融合基因等的连接点在多数情况下广泛地分布于各基因的内含子。因此,在通常的杂交捕获法中,为了捕捉融合基因以及外显子跳跃的连接点,而需要对内含子无偏移地制作探针,需要多个探针。此外,作为用于从新鲜的冷冻样品或细胞株检测融合转录物的替代法,提出了rna测序(rna-seq)。但是,在福尔马林固定石蜡包埋(ffpe)等的rna的质量较低的样品(低质rna样品)中,由于难以通过通常mrna浓缩所使用的多聚a选择等来制作能够信赖的文库,因此较难应用。此外,也有在使用低质rna样品的情况下cdna捕获法或基于锚定多重pcr的方法对rna-seq有用的报告,但由于在这些方法中成为对象的基因的种类非常受限定,因此临床上的有用性较低。因此,一直谋求对于低质rna样品也能够简便地检测多个靶向基因的方法。非专利文献1:j.erikson等人,proc.natl.acad.sci.,usa83,1807-1811,1986非专利文献2:m.soda等人,nature,448,561-566,2007非专利文献3:t.kohno等人,nat.med.18,375-377,2012非专利文献4:k.takeuchi等人,nat.med.18,378-381,2012非专利文献5:d.lipson等人,nat.med.18,382-384,2012非专利文献6:l.fernandez-cuesta等人,cancerdiscov.4,415-422,2014非专利文献7:a.vaishnavi等人,nat.med.,19,1469-1472,2013非专利文献8:r.wang,l等人,clin.cancerres.20,,4107-4114,2014技术实现要素:在一实施方式中,本发明的课题在于提供一种能够简便地检测由融合基因和/或外显子跳跃产生的转录产物的方法。本发明人发现,制作为了检测在大规模并行测序中由基因组上的融合基因或外显子跳跃产生的转录产物而可使用的探针,使用本探针能够有效地检测由基因组上的融合基因或外显子跳跃产生的转录产物。本申请发明包含以下的方式。(1)一种探针,是用于在大规模并行测序中判定基因组上的融合基因的转录产物的存在或其表达量的探针,所述融合基因表达5’侧的基因a的一部分和3’侧的基因b的一部分在假想连接点连接后的转录产物,所述探针与来源于从所述转录产物制备的cdna的基因a或b中的任一方的区域杂交,在将探针与所述cdna杂交时从所述探针的末端部至所述假想连接点的最短碱基长设为x、将所述探针中与cdna杂交的区域的碱基长设为y、将大规模并行测序的读长设为z的情况下,z≥x+y。(2)一种探针组,是用于在大规模并行测序中判定基因组上的融合基因的转录产物的存在或其表达量的探针组,所述融合基因表达5’侧的基因a的一部分和3’侧的基因b的一部分在假想连接点连接后的转录产物,包含至少两个不同的探针,该至少两个不同的探针与来源于从所述转录产物制备的cdna的基因a或b中的任一方的区域杂交,在将探针与所述cdna杂交时从各所述探针的末端部至所述假想连接点的最短碱基长设为x、将在各所述探针中与cdna杂交的区域的碱基长设为y、将大规模并行测序的读长设为z的情况下,z≥x+y。(3)一种探针,是用于在大规模并行测序中判定由外显子跳跃产生的转录产物的存在或其表达量的探针,在所述转录产物中,5’侧的外显子a’和3’侧的外显子b’在假想连接点连接,所述探针与来源于从所述转录产物制备的cdna的外显子a’或b’中的任一方的区域杂交,在将探针与所述cdna杂交时从所述探针的末端部至所述假想连接点的最短碱基长设为x、将所述探针中与cdna杂交的区域的碱基长设为y、将大规模并行测序的读长设为z的情况下,z≥x+y。(4)一种探针组,是用于在大规模并行测序中判定由外显子跳跃产生的转录产物的存在或其表达量的探针组,在所述转录产物中,5’侧的外显子a’和3’侧的外显子b’在假想连接点连接,包含至少两个不同的探针,所述至少两个不同的探针与来源于从所述转录产物制备的cdna的外显子a’或b’中的任一方的区域杂交,在将探针与所述cdna杂交时从各所述探针的末端部至所述假想连接点的最短碱基长设为x、将在各所述探针中与cdna杂交的区域的碱基长设为y、将大规模并行测序的读长设为z的情况下,z≥x+y。(5)根据(1)~(4)中任一项所述的探针或探针组,x为0~140,y为30~140,z为100~300。(6)根据(2)、(4)以及(5)中任一项所述的探针组,包含至少六个所述探针。(7)根据(2)以及(4)~(6)中任一项所述的探针组,仅由满足z≥x+y的探针构成。(8)根据(2)以及(4)~(7)中任一项所述的探针组,探针组包含n个探针,在将各探针的所述最短碱基长分别设为x1、x2、x3、……xn(只是,x1<x2<x3……<xn)的情况下,x1=0、x2=xn×1/(n-1)、x3=xn×2/(n-1)、……xn=xn×(n-1)/(n-1)。(9)一种探针,是用于在大规模并行测序中判定基因组上的融合基因的转录产物的存在或其表达量的探针,所述融合基因表达5’侧的基因a的一部分和3’侧的基因b的一部分在假想连接点连接后的转录产物,与从所述转录产物制备的cdna的包含所述假想连接点的区域杂交。(10)一种探针组,是用于在大规模并行测序中判定基因组上的融合基因的转录产物的存在或其表达量的探针组,所述融合基因表达5’侧的基因a的一部分和3’侧的基因b的一部分在假想连接点连接后的转录产物,包含至少两个不同的探针,该至少两个不同的探针与从所述转录产物制备的cdna的包含所述假想连接点的区域杂交。(11)一种探针,是用于在大规模并行测序中判定由外显子跳跃产生的转录产物的存在或其表达量的探针,在所述转录产物中,5’侧的外显子a’和3’侧的外显子b’在假想连接点连接,所述探针与从所述转录产物制备的cdna中的能够产生外显子跳跃的包含所述假想连接点的区域杂交。(12)一种探针组,是用于在大规模并行测序中判定由外显子跳跃产生的转录产物的存在或其表达量的探针组,在所述转录产物中,5’侧的外显子a’和3’侧的外显子b’在假想连接点连接,包含至少两个不同的探针,该至少两个不同的探针与从所述转录产物制备的cdna中的能够产生外显子跳跃的包含所述假想连接点的区域杂交。(13)一种组合探针组,包含(1)~(12)中任一项所述的不同的多个探针或探针组。(14)根据(1)~(12)中任一项所述的探针或探针组、或(13)所述的组合探针组,进一步地包含至少一个基因表达量测量用探针。(15)根据(1)~(14)中任一项所述的探针、探针组或组合探针组,用于针对来源于经加工处理的活体样品的转录产物使用。(16)一种试剂盒,包含(1)~(15)中任一项所述的探针、探针组或组合探针组。(17)一种对转录产物的存在或其表达量进行判定的方法,该转录产物包含基因组上的融合基因的转录产物,所述方法包含如下工序:从来源于受试者的样品制备转录产物的工序;从所述转录产物制备cdna的工序;对与(1)~(15)中任一项所述的探针、探针组或组合探针组的探针杂交后的靶向cdna进行浓缩的工序;对经浓缩的所述靶向cdna进行基于大规模并行测序的序列解析的工序;以及基于所述序列解析的结果对转录产物的存在或其表达量进行判定的工序,该转录产物包含基因组上的融合基因的转录产物。(18)根据(17)所述的方法,所述判定通过以下的工序来进行:在所述融合基因表达5’侧的基因a的一部分和3’侧的基因b的一部分在假想连接点连接后的转录产物时,在将来源于在假想连接点未产生基因融合的基因a的cdna的读出数设为α、将来源于基因b的cdna的读出数设为β、将来源于在假想连接点产生了基因融合的融合基因的cdna的读出数设为γ的情况下,在0<α或β≤γ的情况下,判定为存在融合基因,在0<γ<α或β的情况下,判定为以低的表达量存在融合基因,在α或β>0、γ=0的情况下,判定为不存在融合基因。(19)一种对转录产物的存在或其表达量进行判定的方法,该转录产物包含由外显子跳跃产生的转录产物所述方法包含如下工序:从来源于受试者的样品制备转录产物的工序;从所述转录产物制备cdna的工序;对与(1)~(15)中任一项所述的探针、探针组或组合探针组的探针杂交后的靶向cdna进行浓缩的工序;对经浓缩的所述靶向cdna进行基于大规模并行测序的序列解析的工序;以及基于所述序列解析的结果对转录产物的存在或其表达量进行判定的工序,该转录产物包含由外显子跳跃产生的转录产物。(20)根据(19)所述的方法,所述判定通过以下的工序来进行:在所述转录产物中5’侧的外显子a’和3’侧的外显子b’在假想连接点连接时,在将来源于在假想连接点未产生基因融合的外显子a’的cdna的读出数设为α’、将来源于外显子b’的cdna的读出数设为β’、将来源于由外显子跳跃产生的转录产物的cdna的读出数设为γ’的情况下,在0<α’或β’≤γ’的情况下,判定为存在由外显子跳跃产生的转录产物,在0<γ’<α’或β’的情况下,判定为以较低的表达量存在由外显子跳跃产生的转录产物,在α’或β’>0、γ’=0的情况下,判定为不存在由外显子跳跃产生的转录产物。(21)根据(17)~(20)中任一项所述的方法,在所述判定工序中,包含以下步骤:在存在与相同区域杂交的多个探针的情况下基于该多个探针的数目来校正转录产物的表达量。(22)根据(17)~(21)中任一项所述的方法,在所述判定工序中,包含以下步骤:基于持家基因的表达量来校正转录产物的表达量。(23)一种判定受试者有无罹患疾病或其风险、确定癌症的种类或判定癌症的预后的方法,所述方法包含如下工序:按照(17)~(21)中任一项所述的方法对转录产物的存在或其表达量进行判定的工序,该转录产物包含基因组上的融合基因的转录产物和/或由外显子跳跃产生的转录产物。(24)根据((23)所述的方法,癌症种类的确定包含以下步骤:基于多个转录产物的存在和/或表达量而对来源于受试者的样品进行聚类。本说明书包含成为本申请的优先权基础的日本专利申请号2017-125074号的公开内容。发明效果通过本发明可提供一种能够简便地检测由融合基因和/或外显子跳跃产生的转录产物的方法。附图说明图1a是本发明的一实施方式的探针的概念图。图示出的探针均是右侧末端部为5’末端,左侧末端部为3’末端。能够以获得包含假想连接点的连接支持读出的方式,基于读长z以及探针中与cdna杂交的区域的碱基长y来确定从探针的末端部至假想连接点的最短碱基长x。图1b示出在本发明的一实施方式的方法中从测序的结果来检测由融合基因和/或外显子跳跃产生的转录产物的方法的一例。如图1b所示,在将来源于在假想连接点未产生基因变异(基因融合或外显子跳跃)的基因a的cdna的读出数设为α、将来源于基因b的cdna的读出数设为β、将来源于在假想连接点产生了基因变异的融合基因的cdna的读出数设为γ的情况下,在0<α或β≤γ的情况下,能够判定为存在变异基因,在0<γ<α或β的情况下,能够判定为以较低的表达量存在变异基因,在α或β>0、γ=0的情况下,能够判定为不存在变异基因。图2a示出所示的各方法中的每10m(1000万)原始读出(rawread)的连接支持读出的数目(泛癌套组(pancancerpanel)表示来源于ffpe的合成cdna的所有外显子捕获)。将使用了本发明的一实施方式的连接捕获法和以往的编码外显子捕获法的情况下的探针数在图2b中示出,将靶向捕获尺寸在图2c中示出。图2b以及图2c中的v1、v2以及v3表示在实施例中所记载的基因套组(panel)(toprnav1、toprnav2以及toprnav3)结果。图3a示出使用三种不同的方法:即,从新鲜冷冻样品提取出的rna的多聚a选择(多聚a捕获)、来源于ffpe的合成cdna的所有外显子捕获(pancancerpanel)、或来源于ffpe的合成cdna的连接捕获,在根据rna-seq而met外显子14跳跃阳性的病例中将测序读出与met转录产物匹配后的结果。图中,由两条纵线夹持的区域表示与met外显子14相当的区域,该区域没有读出表示为外显子跳跃阳性。图3b示出各方法中的每1000万原始读出的met外显子13以及met外显子15的连接(外显子跳跃)支持读出的数目。图4a是苏木精以及伊红染色的骨髓抽吸标本的代表性照片(200×倍率,比例尺100μm)。图4b是苏木精以及伊红染色的tblb标本的代表性照片(左是40×倍率,比例尺1mm。右是400×倍率,比例尺100μm)。图5是示出rna-seq的rpkm、与进行了基于连接捕获法中的覆瓦(tiling)数的校正的rpkm的相关性的图。在a中示出表达量测量用基因群组的结果,在b中示出融合基因解析用基因群组的结果。在所有七个样品中都出现了相关性。图6示出基于基因的表达量将样品聚类后的结果。纵轴表示各基因,根据表达强度进行了聚类。横轴表示各样品,可知根据luad、sarc、muca以及lusc等的癌症类别样品被聚类。具体实施方式1.用于对基因组上的融合基因的转录产物的存在或其表达量进行判定的探针在一方式中,本发明涉及一种用于在大规模并行测序中对基因组上的融合基因的转录产物的存在或其表达量进行判定的探针。在本说明书中,“大规模并行测序(massivelyparallelsequencing)”意思是大规模且并行地实施dna的测序的方法,在大规模并行测序中,通常对102、103、104、105或其以上的分子同时进行测序。大规模并行测序例如包含下一代测序。所谓下一代测序是使用了下一代测序仪的序列信息的获取法,特征在于与sanger法相比能够同时并行地执行庞大数目的测序反应(例如,可参照rickkamps等人.,int.j.mol.sci.,2017,18(2),p.308、以及int.neurourol.j.,2016,20(suppl.2),s76-83)。用于下一代测序的系统被提供各种各样的系统,并不进行限定,例如能够使用roche的genomesequencer(gs)flxsystem(超高通量基因组测序系统)、illumina的hiseq、或genomeanalyzer(ga)(基因组分析仪)、lifetechnologies的supportoligonucleotideligationdetection(solid)(支持寡聚核苷酸连接检测)系统、polonator的g.007系统、以及helicosbiosciences的heliscopegenesequencing(heliscope基因测序)系统等。虽并不进行限定,但在以下示出下一代测序的一般性工序。在下一代测序中,首先最初进行样品制备。在该工序中,结合下一代测序仪的读长,将成为解析对象的核酸酶性或机械性片段化。接着,在多数情况下,附加下一测序工序所需要的接头序列。此外,为了解析特定的基因区域,可以通过pcr等将特定的基因区域富集,或也可以使用探针等对具有特定的序列的区域进行浓缩。基因区域的富集例如能够通过4~12循环的扩增步骤来进行,使用了探针的浓缩能够利用标注于探针的标记(例如生物素等)来进行。接着,进行测序。该工序的详细情况根据下一代测序仪的种类而不同,但典型地是经由接头序列而连接于基板,并将接头序列作为引发部位进行测序反应。对于测序反应的详细情况,例如可参照rickkamps等人(上述)。最后,进行数据输出。在该工序中,获得收集有通过测序反应获得到的序列信息(读出)的信息。能够进一步地解析所输出的数据,导出读出数、例如每个原始读出的连接支持读出数等的更具有意义的结果。在本说明书中,“读出数”是指具有特定的序列的扩增产物的扩增量。由于读出数通常与测序前的核酸的量成比例,因此可利用读出数来推定基因的表达量。在本说明书中,“连接支持读出”意思是包含由基因融合或外显子跳跃产生的转录产物中的连接点、或由基因融合或外显子跳跃产生的基因组上的连接点的读出,“连接支持读出数”意思是连接支持读出的数目。在本说明书中,“原始读出(rawread)”表示通过下一代测序获得到的读出的总数,通过算出每个原始读出的连接支持读出数,从而能够评价连接支持读出的频率。在本说明书中,“基因组上的融合基因”是指由缺失、插入、倒位以及易位等产生的染色体重排导致多个基因连接而产生的变异基因。通常,作为其表达产物,融合基因通过转录产生rna分子。例如,作为rna分子的例子,可列举编码融合蛋白质的mrna等的转录产物。在本说明书中,融合基因的种类并不限定,例如,可列举具有致癌性的融合基因,例如eml4-alk、bcr-abl1、kif5b-ret、slc34a2-ros1、cd74-ros1、ss18-ssx1、ss18-ssx2、nab2-stat6、ewsr1-fli1、syt-ssx1、fus-creb3l2、tpm3-ros1、cd74-nrg1以及ewsr1-fli1等。在本说明中,基因组上的融合基因的转录产物的“存在”是指在基因组上有无融合基因,融合基因的转录产物的“表达量”指来源于融合基因的mrna、rrna以及trna等转录产物、优选mrna的表达量。在一实施方式中,在融合基因表达5’侧的基因a的一部分和3’侧的基因b的一部分在假想连接点连接后的转录产物时,本发明的探针与来源于从上述转录产物制备的cdna的基因a或b的任一方的区域杂交。对于可形成融合基因的各基因以及假想连接点,能够参照科学论文、专利文献以及cosmic等的数据库进行确定。在本说明书中,“外显子”意思是基因的碱基序列中残留在成熟转录产物中的区域。一般而言,在真核生物中,在使基因作为一次转录产物转录之后,通过剪接除去被称为内含子的中间区域,外显子彼此被连接而形成成熟转录产物。例如,在是对蛋白质编码的基因的情况下,产生成熟mirna,该成熟mirna由通过pre-mirna剪接从由转录所产生的mrna前体(pre-mirna)除去内含子而连接的外显子构成。在一实施方式中,在将探针与从转录产物的rna分子制备的cdna杂交时从各探针的5’侧或3’侧中的任一方的末端部至上述假想连接点的最短碱基长设为x、将各上述探针中与cdna杂交的区域的碱基长设为y、将大规模并行测序的读长设为z的情况下,以使z≥x+y的方式来设计探针。以下,也将这样的与不包含假想连接点的核酸区域杂交的探针记载为“不含有假想连接点的探针”。不含有假想连接点的探针具有如下优点:可检测多个融合配偶体以及新融合基因。为了容易理解本发明,在图1a中示出本实施方式的探针的设计。在图1a中,示出从探针的末端部至假想连接点的最短碱基长x、与上述探针中的cdna杂交的区域的碱基长y、以及读长z,示出通过大规模并行测序可获得包含假想连接点的读出。在一实施方式中,读长z由大规模并行测序所使用的设备以及方法等来确定。此外,在来源于样品的核酸片段化的情况下和/或在测序前进行核酸的片段化的情况下,可以通过这些片段的长度规定读长。读长z的长度并不限定,例如可以在50以上、75以上、100以上、150以上或160以上,可以在500以下、400以下、300以下、200以下或180以下,例如可以为50~500、100~300或150~200。另外,在大规模并行测序中,存在仅从核酸的单侧进行测序的单端读出和从核酸的两侧进行测序的双端,但上述读长z优选是双端中的读长。只要是本领域内技术人员则能够适当地确定在探针中与cdna杂交的区域的碱基长y。y例如可以在20以上、30以上、40以上,优选在50以上、60以上或80以上,可以在220以下、200以下、180以下,优选在160以下、140以下或120以下,例如可以为20~220、50~160或60~140。优选探针在自接近假想连接点的末端部连续的区域与cdna杂交。在一实施方式中,探针在其全长中与cdna杂交,在该情况下,y与探针的长度变为相同。探针的碱基长并不限定,例如可以在20以上、40以上、60以上、80以上、100以上、110以上或115以上,可以在220以下、200以下、180以下、160以下、140以下、130以下或125以下,例如可以为20~220、60~180、100~140、110~130、115~125或120。基于上述读长z以及在探针中与cdna杂交的区域的碱基长y,能够适当地确定从探针的末端部至假想连接点的最短碱基长x。例如,从探针的末端部至假想连接点的最短碱基长x的下限为零,其意思是针对邻接于假想连接点的区域设计探针。x的上限并不限定,例如可以在300以下、250以下、200以下、150以下、140以下、130以下、125以下、120以下,x例如可以为0~300、0~200、0~140、0~125或0~120。z≥x+y+a(a≥0)表示可获得包含超过假想连接点a碱基以上的序列的读出,这样,通过在假想连接点的附近设计多个探针,从而可使用该探针有效地浓缩与融合基因相关的各种各样的种类的转录产物。a的值只要在0以上则并不限定,但由于当增大时特异性会变高而检测灵敏度下降,因此只要是本领域内技术人员则能够参照本说明书的内容适当地确定。a的值例如可以在5以上、10以上,优选在15以上、20以上、30以上、50以上或100以上,可以在500以下、400以下,优选在300以下、200以下或150以下。只要是本领域内技术人员则能够基于靶向基因的序列容易地设计探针。在本说明书中,“靶向基因”是指可通过本发明的探针捕捉的基因,例如能够形成融合基因的基因以及能够产生外显子跳跃的基因等。作为这样的探针的例子,例如,可列举包含如下碱基序列的探针:(a)靶向基因的互补性序列连续的至少20、40、60、80、100、110、115或120的碱基序列;(b)在(a)的碱基序列中附加、缺失和/或置换一个或多个碱基后的碱基序列;(c)相对于(a)的碱基序列,具有例如70%以上、80%以上、优选90%以上、95%以上、97%以上、98%以上或99%以上的同一性的碱基序列;以及(d)在严格条件下,针对靶向基因连续的至少20、40、60、80、100、110、115或120的序列进行杂交的核酸的碱基序列。在本说明书中,“一个或多个”的范围是一到十个,优选为一到七个,进一步地优选为一到五个,特别是优选为一到三个或者一个或两个。此外,在本说明书中,与碱基序列相关的同一性的值,表示使用对多个序列间的同一性进行运算的软件(例如,fasta、danasys以及blast)按默认的设定算出的值。对于同一性的确定方法的详细情况,例如,可参照altschul等人,nuc.acids.res.25,3389-3402,1977以及altschul等人,j.mol.biol.215,403-410,1990。在本说明书中,“严格条件”意思是形成所谓的特异性杂交而不形成非特异性杂交的条件。严格条件能够利用公知的杂交法的条件。例如,只要参照greenandsambrook,molecularcloning(分子克隆),4thed(2012),coldspringharborlaboratorypress(美国冷泉港实验室期刊)适当地确定即可。具体而言,只要通过杂交法温度、溶液所包含的碱浓度、以及杂交法的清洗工序中的温度、溶液所包含的碱浓度来设定严格条件即可。作为更详细的严格条件,例如,可列举钠浓度为25~500mm,优选为25~300mm,温度为42~68℃,优选为42~65℃。更具体而言,可列举5×ssc(83mmnacl、83mm柠檬酸钠)、温度42℃。能够基于上述序列通过本领域内技术人员所知的公知的方法来制备探针,并不被限定,例如,能够通过化学合成法来制备。在一实施方式中,本发明涉及包含至少两个不同的上述探针的探针组。上述探针的数目只要在两个以上则并不特别限定,但由于如果过少则检测灵敏度降低,而当过多时成本会增加,因此只要考量灵敏度以及成本等并参照本说明书的内容适当地确定即可。可包含于探针组的上述探针的数目例如可以在三个以上、四个以上、五个以上、六个以上、八个以上、十个以上或十一个以上,此外,可以在三十个以下、二十五个以下、二十个以下、十五个以下、十四个以下、十三个以下或十二个以下。优选探针组所包含的各探针从末端部至上述假想连接点的最短碱基长x的各个值并不相同,并是分散的。是因为,由此可捕捉各种各样的核酸片段。例如,探针组包含n个探针,在将各探针的最短碱基长分别设为x1、x2、x3、……xn(只是,x1<x2<x3……<xn)的情况下,能够以x1=0±b、x2=xn×1/(n-1)±b、x3=xn×2/(n-1)±b、…xn=xn×(n-1)/(n-1)±b的方式确定各探针的最短碱基长。b是常数,b为零的情况下,意味着各探针的最短碱基长x自假想连接点均匀地分布,b的值越大则意味着自假想连接点的分布越不均匀。b例如在50以下、40以下、30以下、25以下、20以下、15以下、10以下,优选在5以下、4以下、3以下、2以下、1以下或0。此外,xn可以是任意值,例如可以为20~500、30~400、40~300、60~200、80~180,优选为100~140、110~130、115~125或120。此外,在探针的数目n为3以上的情况下,也可以在按照上述式设计探针之后,从探针组去掉m个探针(只是,m为1以上的整数,例如为1~5、1~4、1~3、1~2,优选为1,n-m≥2)。在一实施方式中,为了在下一代测序的测序工序之前浓缩特定的核酸序列而能够使用本发明的探针。在一方式中,本发明的探针与包含假想连接点的核酸区域杂交。也将这样的与包含假想连接点的核酸区域杂交的探针记载为“含有假想连接点的探针”。对于含有假想连接点的探针或其组,包含与包含假想连接点的核酸区域杂交的探针以外的构成、例如在探针中与cdna杂交的区域的碱基长y以及探针组所包含的探针的数目,与上述的“不含有假想连接点的探针”是同样的。只是,由于含有假想连接点的探针仅检测由基因a的一部分和基因b的一部分的融合而产生的一个融合基因,因此特异性较高,但无法检测各种各样的融合配偶体。在一实施方式中,含有假想连接点的探针与来源于从上述融合基因的转录产物制备的cdna的5’侧的基因a和3’侧的基因b双方的区域的10碱基以上、15碱基以上、20碱基以上、30碱基以上、40碱基以上、50碱基以上或60碱基以上杂交。在一实施方式中,本发明的探针组除了上述“不含有假想连接点的探针”之外,还包含“含有假想连接点的探针”。通过包含两者的探针,从而可进一步地提高检测特异性。在一实施方式中,本发明的探针组仅由满足z≥x+y的探针以及上述含有假想连接点的探针构成。此外,在另一实施方式中,本发明的探针组仅由满足z≥x+y的探针构成。也可以对成为评价对象的所有靶向基因的外显子的5’末端侧以及3’末端侧设计本发明的探针组,但优选仅对已知形成融合基因的基因的、与融合基因关联的外显子的5’末端侧和/或3’末端侧设计探针。在一实施方式中,本发明的探针或探针组进一步地包含至少一个基因表达量测量用探针。基因表达量测量用探针是为了在大规模并行测序中测量基因表达量而使用的探针。针对将要测量表达量的基因,能够无遗漏地、或者例如以2×覆瓦(tiling)以上的密度来设计基因表达量测量用探针。基因表达量测量用探针的碱基长并不限定,例如可以在20以上、40以上、60以上、80以上、100以上、110以上或115以上,可以在220以下、200以下、180以下、160以下、140以下、130以下或125以下,例如可以为20~220、60~180、100~140、110~130、115~125或120。针对一个基因的基因表达量测量用探针的数目并不限定,例如可以在三个以上、四个以上、五个以上、六个以上、八个以上、十个以上或十一个以上,此外,可以在三十个以下、二十五个以下、二十个以下、十五个以下、十四个以下、十三个以下或十二个以下。基因表达量测量用探针可以是针对“多个”基因、例如2以上、5以上、10以上、50以上、100以上、150以上、200以上、250以上、优选300以上、400以上或500以上、可以为2000以下、1000以下、900以下、优选800以下、700以下或600以下的基因的探针。作为测量表达量的靶向基因的例子,可列举癌症基因(例如,alk、egfr、erbb2、met)以及持家基因。能够将可与这些基因的至少一部分结合的核酸作为探针使用。通过包含基因表达量测量用探针,从而能够更准确地测量基因的表达量。在一实施方式中,本发明涉及一种包含不同的多个上述探针或探针组的组合探针组。在此,优选组合探针组包含针对多个不同的融合基因的探针组,由此可同时检测多个融合基因的转录产物存在或其表达量。“多个”的下限以及上限并不特别限定,例如可以在2以上、5以上、10以上、50以上、100以上、150以上、200以上、250以上,优选在300以上、400以上或500以上,可以在2000以下、1000以下、900以下,优选在800以下、700以下或600以下。在一实施方式中,优选地,针对所包含的rna分解或劣化后的样品、例如来源于进行了加工处理的活体样品的转录产物,使用本发明书所记载的探针、探针组或组合探针组。作为加工处理的种类,可列举热处理、冷冻处理、酸处理、碱处理,优选ffpe(福尔马林固定石蜡包埋)等的固定处理等。2.本发明的探针的效果正如上述那样,由于本发明的探针能够捕捉并浓缩通过大规模并行测序可获得包含假想连接点的读出之类的核酸片段,因此通过针对进行了浓缩的样品进行大规模并行测序,从而可有效地检测融合基因。此外,在一实施方式中,由于本发明的探针组针对从mrna等的转录产物制备的cdna使用,此外在假想连接点附近可集中地包含探针,因此可具有如下优点:比起捕捉基因组dna的内含子部分的内含子捕获法、以及捕捉所有外显子部分的编码外显子捕获法,所需要的探针的数目较少。此外,在一实施方式中,由于本发明的探针组在假想连接点附近集中地包含探针,因此可获得包含假想连接点的各种各样的核酸片段。根据ryantewhey等人(genomebiology,2009,10,r116),显示即使将探针设为2×覆瓦以上的密度coverage也不增高,因此令人惊讶的是,通过在假想连接点附近集中地包含探针,从而融合基因或外显子跳跃的检测效率增高。另外,在本说明书中,“覆瓦”意思是针对靶向基因设计探针的密度,覆瓦的倍数的值n意思是当将探针的长度设为w时逐个隔开w/n的间隔来设计探针。此外,在一实施方式中,由于转录或浓缩不需要mrna所包含的多聚a序列,因此本发明的探针特别是在rna分解或劣化后的样品中可有效地检测融合基因。3.用于对由外显子跳跃产生的转录产物的存在或其表达量进行判定的探针在一方式中,本发明涉及一种探针或包含至少两个不同的本探针的探针组,其用于在大规模并行测序中对由外显子跳跃产生的转录产物的存在或其表达量进行判定。在上述转录产物中5’侧的外显子a’和3’侧的外显子b’在假想连接点被连接的情况下,本方式的探针与来源于从上述转录产物制备的cdna的外显子a’或b’中的任一方的区域杂交。在一实施方式中,在将探针与从上述转录产物制备的cdna杂交时从各探针的末端部至上述假想连接点的最短碱基长设为x、将在各上述探针中与cdna杂交的区域的碱基长设为y、将大规模并行测序的读长设为z的情况下,z≥x+y。在一方式中,涉及一种探针或包含至少两个不同的本探针的探针组,该探针是用于在大规模并行测序中对由外显子跳跃产生的转录产物的存在或其表达量进行判定的探针,在上述转录产物中5’侧的外显子a’和3’侧的外显子b’在假想连接点连接,所述探针与从上述转录产物制备的cdna中的能够产生外显子跳跃的包含假想连接点的区域杂交。在本说明书中,“外显子跳跃”是指如下现象:由于剪接错误而使一部分外显子与内含子一起被除去,结果产生异常的外显子连接。例如,相当于如下情况:在野生型基因包含外显子a’、b’以及c’的情况下,原本应该外显子a’、b’以及c’被连接的位置,由于剪接的错误而跳过外显子b’,使外显子a’和外显子c’被连接。由于由外显子跳跃产生的产物为以上产物,因此较多成为疾病的原因。例如,已知met(mesenchymal-epithelialtransition:上皮间充质转化)的外显子14的跳跃与非小细胞肺癌的罹患率具有关联。作为用于对由外显子跳跃产生的转录产物的存在或其表达量进行判定的探针以外的本方式的探针的构成,例如探针的数目、从各探针的末端部至假想连接点的最短碱基长x、与各探针中的cdna杂交的区域的碱基长y、大规模并行测序的读长z、各探针的序列、设计等,依据上述“1.用于对基因组上的融合基因的转录产物的存在或其表达量进行判定的探针”。对于可进一步地包含基因表达量测量用探针的方面,与“1.用于对基因组上的融合基因的转录产物的存在或其表达量进行判定的探针”也是同样的。此外,本方式的探针的效果依据上述“2.本发明的探针的效果”。在一方式中,本发明涉及一种探针组,其包含上述“1.用于对基因组上的融合基因的转录产物的存在或其表达量进行判定的探针”、以及本方式的“用于对由外显子跳跃产生的转录产物的存在或其表达量进行判定的探针”两者。通过使用该探针组,可同时检测融合基因和外显子跳跃两者。4.包含探针的试剂盒在一方式中,本发明涉及一种试剂盒,其包含上述“1.用于对基因组上的融合基因的转录产物的存在或其表达量进行判定的探针”和/或上述“3.用于对由外显子跳跃产生的转录产物的存在或其表达量进行判定的探针”所记载的探针、探针组或组合探针组。本试剂盒除了上述探针之外,例如还可以包含缓冲液、酶以及使用说明书等。为了对融合基因的转录产物的存在或其表达量、和/或由外显子跳跃产生的转录产物的存在或其表达量进行判定,可使用本试剂盒。5.对包含融合基因的转录产物的转录产物的存在或其表达量进行判定的方法在一方式中,本发明涉及一种对包含基因组上的融合基因的转录产物的转录产物的存在或其表达量进行判定的方法。本方式的方法按如下顺序包含:从来源于受试者的样品制备转录产物的工序(转录产物制备工序);从转录产物制备cdna的工序(cdna制备工序);对与上述“1.用于对基因组上的融合基因的转录产物的存在或其表达量进行判定的探针”的探针、探针组或组合探针组的探针杂交后的靶向cdna进行浓缩的工序(浓缩工序);对经浓缩的靶向cdna进行基于大规模并行测序的测序的工序(测序工序);以及基于测序的结果对包含基因组上的融合基因的转录产物的存在或其表达量进行判定的工序(判定工序)。以下,详细地对构成本方法的各工序进行说明。(1)转录产物制备工序在转录产物制备工序中,从来源于受试者的样品制备转录产物。在本说明书中,受试者的物种并不限定,优选哺乳动物,例如人以及黑猩猩等灵长类,大鼠以及小鼠等实验动物,猪、牛、马、绵羊以及山羊等家畜动物、以及狗和猫等宠物,优选人。在本说明书中,“样品”意思是供给于本发明的方法的活体试样。作为能够在本发明中使用的样品,并不限定,例如,可列举从活体离析出的体液、细胞或组织。作为体液的例子,可列举血液、汗、唾液、乳汁以及尿等,作为细胞的例子,例如,可列举末梢血细胞、包含细胞的淋巴液以及组织液、毛母细胞、口腔细胞、鼻腔细胞、肠道细胞、阴道细胞、粘膜细胞、痰(可包含肺泡细胞或肺泡壁细胞等)。作为组织的例子,可列举癌症的病变部位,例如脑、咽、甲状腺、肺、乳房、食道、胃、肝脏、胰腺、肾脏、小肠、大肠、膀胱、前列腺、子宫、卵巢,优选肺等,例如能够使用这些组织的活检样品。在使用活检样品的情况下,由于能够同时进行组织学上的病理诊断和基于本发明的方法的融合基因的检测,因此能够更准确地确定受试者的病理学上的症状。此外,在一实施方式中,使用所包含的rna分解或劣化后的样品、例如进行了加工处理的活体样品来作为样品。作为加工处理的种类,例如,可列举热处理、冷冻处理、酸处理、碱基处理,优选ffpe(福尔马林固定石蜡包埋)等的固定处理等。转录产物(总rna)可包含rrna、trna以及mrna,但优选mrna。来自样品的转录产物的制备能够使用公知的任意方法来进行。例如,能够将样品与包含硫氰酸胍以及表面活性剂的可溶化液进行混合,对获得到的混合液实施物理性处理(搅拌、均质、超声波粉碎等),并提取转录产物。优选的是,进一步地也能够利用如下方法(agpc法):添加苯酚以及氯仿并搅拌,并通过对其进行离心分离,从而回收包含转录产物的水层。接着,能够通过醇沉淀法等从该水层获得转录产物。此外,也能够对rna的提取使用rna-bee(tel-testinc.)以及trizol(thermofisherscientific:赛默飞世尔科技)等的市售的试剂盒。对于这些具体性操作等,只要参照该领域的协议、例如greenandsambrook,molecularcloning,4thed(2012),coldspringharborlaboratorypress即可。对于本说明书所记载的其他生物学上的方法、例如以下的cdna制备工序以及浓缩工序,也能够参照greenandsambrook(上述)。(2)cdna制备工序来自在转录产物制备工序中获得到的转录产物的cdna的制造能够通过使用逆转录酶的逆转录反应来进行。在进行逆转录反应时使用的引物、逆转录酶以及反应条件等,只要是本领域内技术人员则能够适当地选择公知的条件。在本发明的方法中,由于通过在以下记载的浓缩工序使目标的核酸片段浓缩,因此不需要利用多聚a序列仅使mrna逆转录,例如,也可以使用随机引物等使总rna逆转录。(3)浓缩工序在浓缩工序中,对与本说明书所记载的探针、探针组或组合探针组杂交后的靶向cdna进行浓缩。能够使用本领域内技术人员所知的任意方法进行浓缩。例如,对探针标注标记,并通过标记与其他物质的相互作用,能够将与探针杂交后的靶向cdna浓缩。例如,可以对探针标注生物素,并通过与抗生物素蛋白的相互作用来浓缩与探针杂交后的靶向cdna,可以通过利用了基质或抗原抗体反应的亲和层析来进行浓缩,也可以对探针标注磁性微珠,并通过磁力来浓缩与探针杂交后的cdna。也可以在基于探针组的浓缩工序之前或之后,结合大规模并行测序中的读长,将cdna酶性或机械性片段化。此外,也可以附加下一测序工序所需要的接头序列。为了在浓缩工序之前或之后解析特定的基因区域,也可以通过pcr等将特定的基因区域富集。基因区域的富集例如能够通过4~12循环的扩增步骤来进行。(4)测序工序在测序工序中,对经浓缩的靶向cdna进行基于大规模并行测序的测序。测序工序的详细情况根据大规模并行测序所使用的设备的种类等而不同,但典型地是经由接头序列而连接于基板,此外,将接头序列作为引发部位进行测序反应。对于测序反应的详细情况,例如可参照rickkamps等人(上述)。在本工序中,可获得收集有通过测序反应获得到的序列信息(读出)的信息。能够进一步地解析所输出的数据,导出读出数、例如每个原始读出的连接支持读出数等的更具有意义的结果。在各厂商中有市售用于大规模并行测序的装置,能够利用它们。例如,并不进行限定,能够使用roche的genomesequencer(gs)flxsystem、illumina的hiseq、或genomeanalyzer(ga)、lifetechnologies的supportoligonucleotideligationdetection(solid)系统、polonator的g.007系统、以及helicosbiosciences的heliscopegenesequencing系统等。(5)判定工序在判定工序中,基于测序工序的结果对包含基因组上的融合基因的转录产物的存在或其表达量进行判定。在图1b中示出判定工序的一例。判定工序的具体性方法并不限定,例如能够根据以下的基准来进行。在融合基因表达5’侧的基因a的一部分和3’侧的基因b的一部分在假想连接点连接后的转录产物时,在将来源于在假想连接点未产生基因融合的基因a的cdna的读出数设为α、将来源于基因b的cdna的读出数设为β、将来源于在假想连接点产生了基因融合的融合基因的cdna的读出数设为γ的情况下,在0<α或β≤γ的情况下,能够判定为存在融合基因,在0<γ<α或β的情况下,能够判定为以较低的表达量存在融合基因,在α或β>0、γ=0的情况下,能够判定为不存在融合基因。在α和/或β=0以及γ=0的情况下,可认为是不存在融合基因的转录产物、或由于样品的质量不好而转录产物被分解的任一方。在该情况下,通过对推定融合基因的两方的基因的野生型转录产物的假想连接点附近的读出更详细地计数,从而能够准确地判定哪一方正确。由于读出数通常与测序前的核酸的量成比例,因此能够基于基因的读出数来判定表达量。表达量例如能够通过与野生型基因的读出数的比较、与健康体中的读出数的比较等而以相对值来确定,也能够将特定的条件下的读出数等的测量值作为绝对值来确定。在一实施方式中,上述判定工序包含在存在与相同区域杂交的多个探针的情况下基于该多个探针的数目来校正转录产物的表达量。由于本发明的探针组在假想连接点附近集中地包含探针,因此可与相同区域重复地设计探针。与此相伴,根据探针的数目,可较高地算出与该区域相当的转录产物的读出数。因此,为了基于读出数更准确地判定表达量,优选通过与相同区域杂交的探针的数目来校正读出数。基于探针的数目的读出数的校正方法并不限定,例如,通过用读出数除以探针的覆瓦数,从而能够校正读出数(例如,如果是5×覆瓦,则能够用读出数除以5,如果是10×覆瓦,则能够用读出数除以10)。在一实施方式中,上述判定工序包含基于至少一个持家基因的表达量来校正转录产物的表达量。基于持家基因的校正,在使用不同的探针组的情况下、和/或使用不同的样品的情况下,在更准确地比较表达量时特别优选。持家基因能够使用在本领域公知的基因,例如能够使用actb、b2m、gapdh、gusb、h3f3a、hprt1、hsp90ab1、npm1、ppia、rplp0、tfrc以及ubc中的至少一个、至少两个、至少三个、至少五个或所有。基于持家基因的读出数的校正方法并不限定,例如,通过用将要测量表达量的转录产物的读出数除以持家基因的读出数,从而能够校正读出数。根据本方式的方法,通过判定基因组上的融合基因存在或其表达量而可诊断疾病。此外,根据基因组上的融合基因存在或其表达量的信息等的受试者的遗传上的背景,可选择合适的药剂等的疗法。6.对包含由外显子跳跃产生的转录产物的转录产物的存在或其表达量进行判定的方法在一方式中,本发明涉及一种对包含由外显子跳跃产生的转录产物的转录产物的存在或其表达量进行判定的方法。本方式的方法按如下顺序包含:从来源于受试者的样品制备转录产物的工序(转录产物制备工序);从转录产物制备cdna的工序(cdna制备工序);对与上述“3.用于对由外显子跳跃产生的转录产物的存在或其表达量进行判定的探针”所记载的探针、探针组或组合探针组的探针杂交后的靶向cdna进行浓缩的工序(浓缩工序);对经浓缩的靶向cdna进行基于大规模并行测序的测序的工序(测序工序);以及基于测序的结果对包含由外显子跳跃产生的转录产物的转录产物的存在或其表达量进行判定的工序(判定工序)。除了用于对包含由外显子跳跃产生的转录产物的转录产物的存在或其表达量进行判定方面、以及所使用的探针不同方面以外的本方式的方法的构成,例如转录产物制备工序、cdna制备工序、浓缩工序、测序工序、判定工序,依据上述“5.对融合基因的转录产物的存在或其表达量进行判定的方法”。因此,在此,以与上述“5.对融合基因的转录产物的存在或其表达量进行判定的方法”不同的方面为中心,在以下进行说明。在一方式中,本发明涉及一种使用上述“1.用于对基因组上的融合基因的转录产物的存在或其表达量进行判定的探针”、以及上述“3.用于对由外显子跳跃产生的转录产物的存在或其表达量进行判定的探针”两者来进行cdna浓缩工序的方法。由此,可同时检测融合基因和外显子跳跃两者。能够正如在上述“5.对融合基因的转录产物的存在或其表达量进行判定的方法”中所记载的那样来进行判定工序。即,能够通过如下的工序来进行:在转录产物中5’侧的外显子a’和3’侧的外显子b’在假想连接点连接时,在将来源于在假想连接点未产生基因融合的外显子a’的cdna的读出数设为α’、将来源于外显子b’的cdna的读出数设为β’、将来源于由外显子跳跃产生的转录产物的cdna的读出数设为γ’的情况下,在0<α’或β’≤γ’的情况下,判定为存在由外显子跳跃产生的转录产物,在0<γ’<α’或β’的情况下,判定为以较低的表达量存在由外显子跳跃产生的转录产物,在α’或β’>0、γ’=0的情况下,判定为不存在由外显子跳跃产生的转录产物。7.判定有无罹患疾病或其风险、确定癌症的种类或判定癌症的预后的方法在一方式中,本发明涉及一种判定受试者有无罹患疾病或其风险、确定癌症(例如,原发癌症)的种类或判定癌症(或癌症患者)的预后的方法,该方法包含按照本说明书所记载的方法对包含基因组上的融合基因的转录产物、和/或由外显子跳跃产生的转录产物的转录产物的存在或其表达量进行判定的工序(判定工序)。能够正如在上述“5.对融合基因的转录产物的存在或其表达量进行判定的方法”、和/或在上述“6.对由外显子跳跃产生的转录产物的存在或其表达量进行判定的方法”中所记载的那样来进行判定工序。本方式的方法在判定有无罹患疾病或其风险、确定癌症的种类或判定癌症的预后的方面,与上述“5.对融合基因的转录产物的存在或其表达量进行判定的方法”、或“6.对由外显子跳跃产生的转录产物的存在或其表达量进行判定的方法”所记载的方法不同。在本方式的方法中,只要疾病的种类能够通过融合基因或外显子跳跃来判定有无罹患或其风险,则并不限定,例如,可列举脑肿瘤、咽癌、甲状腺癌、肺癌、乳腺癌、食道癌、胃癌、肝癌、胰腺癌、肾癌、小肠癌、大肠癌、膀胱癌、前列腺癌、宫颈癌、卵巢癌、肉瘤、淋巴瘤或黑色素瘤,优选肺癌或肉瘤。本方式的方法除了判定工序之外,还可包含如下工序:基于基因组上的融合基因的转录产物的存在或其表达量、和/或由外显子跳跃产生的转录产物的存在或其表达量来评价受试者有无罹患疾病或其风险的工序(评价工序);确定癌症的种类的工序(确定工序);或判定癌症的预后的工序(判定工序)。评价工序能够利用融合基因或外显子跳跃与疾病的公知的关联性来进行评价工序。例如,eml4(echinodermmicrotubuleassociatedproteinlike4,棘皮动物微管相关样蛋白4)-alk(anaplasticlymphomakinase,间变性淋巴瘤激酶)能够用于判定有无罹患非小细胞肺癌或其风险,bcr(bcellreceptor,b细胞受体)-abl1(abelsonmurineleukemiaviraloncogenehomolog1,阿贝尔森鼠白血病病毒致癌基因同源物1)能够用于判定有无罹患慢性髓细胞白血病或其风险,taf15(tata-boxbindingproteinassociatedfactor15,tata盒结合蛋白相关因子15)-nr4a3(nuclearreceptorsubfamily4groupamember3,核受体亚家族4组a成员3)能够用于判定有无罹患骨外软骨肉瘤或其风险,ahrr(aryl-hydrocarbonreceptorrepressor,芳烃受体阻遏物)-ncoa2(nuclearreceptorcoactivator2核受体共激活子2)能够用于判定有无罹患血管纤维瘤或其风险,met的外显子14的跳跃能够用于判定有无罹患非小细胞肺癌或其风险。在评价工序中,在检测到融合基因的转录产物的存在或由外显子跳跃产生的转录产物的存在的情况下,或者在融合基因的表达量或由外显子跳跃产生的转录产物的表达量例如与健康体相比高的情况下,能够评价为罹患了该疾病或其风险较高。确定工序以及判定工序能够利用包含基因组上的融合基因的转录产物、和/或由外显子跳跃产生的转录产物的转录产物与疾病的关联性来进行癌症的种类的确定以及癌症的预后的判定。上述转录产物与疾病的关联性可以利用公知的关联性,也可以利用未知的关联性。在本说明书中,“预后”意思是例如进行了化学疗法等的治疗处置之后的、肿瘤量的减少、肿瘤增殖的抑制、疾病的经过或结局(例如,有无复发、生死等),优选是生存期限的长短、复发的风险的高低。预后的判定例如可以是进行了治疗处置之后的、生存期限或一定期限后的生存率的预测。在一实施方式中,确定工序以及判定工序包含基于多个转录产物的存在和/或表达量而对来源于受试者的样品进行聚类。该实施方式在上述转录产物与疾病的关联性为未知的情况下特别有利。该实施方式中的多个转录产物的数目并不限定,例如可以在2以上、5以上、10以上、20以上、30以上、50以上、100以上、200以上、300以上、400以上或500以上,可以在20000以下、10000以下、5000以下,优选在3000以下、2000以下或1000以下。在基于多个转录产物的存在和/或表达量对样品进行聚类时,能够加上来源于癌症类别已确定或预后已被预测的受试者的标准样品。由此,能够更准确地进行基于癌症类别或预后的聚类。聚类的方法并不限定,例如能够使用统计解析软件r的heatmap.3,并以基因表达量为基础来进行样品的聚类。确定工序中的癌症的种类并不限定,例如可以是脑肿瘤、咽癌、甲状腺癌、肺癌(例如,肺腺癌)、乳腺癌、食道癌、胃癌、肝癌、胰腺癌、肾癌、小肠癌、大肠癌、膀胱癌、前列腺癌、宫颈癌、卵巢癌、肉瘤、淋巴瘤或黑色素瘤,优选肺癌(例如,肺腺癌)或肉瘤。本方式的判定有无罹患疾病或其风险、确定癌症的种类或判定癌症的预后的方法也可以与其他方法、例如组织学上的病理诊断、基于fish、rt-pcr以及免疫组织化学等的生物标识的检测、ct、mri以及核医学检查等的图像诊断组合进行。通过与其他方法的组合而能够提高疾病的检测精度。实施例材料和方法gdna靶向测序通过genereaddnaffpekit(qiagen)从ffpe样品离析出基因组dna(500ng),并使用sureselectxtcustomkit(agilent)将靶向片段进行了富集。定制的探针设计为与靶向基因的gdna杂交并进行捕获。通过双端选项,使用hiseq2500platform(illumina)进行了离析出的片段的大规模并行测序(massivelyparallelsequencing)。对于各碱基,从较大的数据集仅选择q值≥20的测序读出,并使用bowtie2算法(http://bowtie-bio.sourceforge.net/bowtie2/index.shtml)与参照人基因组测序(hg19)匹配。根据mutect(http://www.broadinstitute.org/cancer/cga/mutect)确定了体细胞变异。此外,根据以下的判断基准:judgment=keep(keep表示基于mutect的体细胞变异阳性)、肿瘤读出深度≥20×、变异率≥10%以及正常读出深度≥10×,选择出变异候选。基于多聚a选择的rna-seq使用rna-bee(tel-testinc.,#cs-104b)从新鲜冷冻样品提取所有rna,在通过dnasei(lifetechnology)进行处理之后供给于多聚a-rna选择,并将其使用于cdna合成。使用nebnextultradirectionalrnalibraryprepkit(newenglandbiolabs),按照制造厂家的协议进行了rna-seq的文库制备。使用hiseq2500platform(illumina),从各簇(cluster)的两端进行了ngs测序。基于cdna捕获的rna-seq通过rneasyffpekit(qiagen)从ffpe样品提取所有rna,并通过dnasei(lifetechnology)进行了处理。使用trusightrnapan-cancerpanel(illumina),按照制造厂家的协议进行了用于编码外显子捕获的cdna合成、基于探针的捕获以及文库制备。使用sureselectrnacapturekit(agilenttechnologies),按照制造厂家的协议进行了用于连接捕获的cdna合成以及文库制备。用于连接捕获法的客制化探针设计为与靶向基因的假想连接点附近序列杂交并进行捕获。具体而言,考虑到所使用的大规模并行测序的读长为170bp,并且假设在探针中与cdna杂交的区域的碱基长只要在50以上则可获得包含假想连接点的读出,以使探针与cdna杂交时从各探针的末端部至假想连接点的最短碱基长为120以下的方式来设计探针。此外,探针的长度均设为120bp。此外,在连接捕获法中,为了获得尽量多的种类的读出,以5×覆瓦或10×覆瓦进行了探针的设计。使用hiseq2500platform(illumina),从各簇的两端进行了ngs测序。作为一例,在以下的表1中示出为了鉴定eml4的外显子13、alk的外显子20以及eml4-alk的融合基因而所使用的探针组的序列号。[表1]序列号eml4(外显子13)1eml4(外显子13)探针12eml4(外显子13)探针23eml4(外显子13)探针34eml4(外显子13)探针45eml4(外显子13)探针56alk(外显子20)7alk(外显子20)探针18alk(外显子20)探针29alk(外显子20)探针310alk(外显子20)探针411alk(外显子20)探针512实施例1:基于连接捕获法的融合基因的检测结果在测序数据的解析中,对支持融合转录产物的连接点的存在的测序读出的数目进行计数,并与野生型基因的转录产物比较,调查了是否显著地表达融合转录产物。此外,在存在各基因转录产物而不存在融合基因转录产物的情况下,表示不存在融合转录产物,但在各基因的读出数为零的情况下,慎重评价是缘于mrna未表达还是基于样品质量的mrna分解。作为中试实验,基于连接捕获法制作了将六十七个融合基因作为靶向的较小的靶向套组(toprnav1)。然后,将toprnav1与由检测作为以往方法的融合基因的基因组中的连接点的内含子捕获法(topdna)而获得的套组、或基于编码外显子捕获法的trusightrnapan-cancerpanel(illumina)进行了比较。其结果,由连接捕获法获得到的toprnav1套组比起由内含子捕获法获得到的topdna套组能够更准确地检测融合基因,此外,连接支持读出/1000万原始读出的值也多(表2、图2a)。其结果表明了连接捕获法是检测融合基因的优异的方法。[表2]表中,nsclc指非小细胞肺癌,ss指滑膜肉瘤(synovialsarcoma),lgfs指低度恶性纤维黏液样肉瘤(low-gradefibromyxoidsarcoma),arsm指齿槽横纹肌肉瘤(alveolarrhabdomyosarcoma),ews指尤文氏肉瘤(ewingsarcoma)。接着,对于连接捕获法,设计了覆盖肉瘤的融合基因的更大的靶向套组(toprnav2)、以及覆盖在数据库cosmic中报告过的所有融合基因的套组(toprnav3)。提取出rna的ffpe保存样品的rna完整性评分(rnaintegrityscore)(rin)是表示发生了高度分解的1.1~2.3,但能够检测所有融合转录产物(表3)。此外,在连接捕获法中,与通过编码捕获法设计了套组的情况相比,预想的探针的数目以及靶向捕获尺寸(由探针捕捉的核酸序列的长度)两者明显少(图2b以及图2c)。其表明了连接捕获法的性价比非常高。通过计算持家基因覆盖度以及覆盖率,从而能够评价rna-seq的质量。将以下的基准设为rna-seq的质量优异:持家基因的平均覆盖度>500x以及100x、以及持家基因的覆盖率>70%。在不存在连接支持读出的情况下,也存在如下可能性:来源于ffpe的rna发生了分解,从而检测不到连接支持读出。因此,为了确定融合基因真的是阴性,研发出了对在cosmic数据库中报告过的推定融合基因两者的基因的野生型转录产物的连接支持读出进行计数的管道。通过针对病例#31(eml4-alk阳性肺腺癌)的该分析的结果,从而确认了该肿瘤对于分析的融合转录产物真的是阴性(未示出数据)。实施例2:基于连接捕获法的外显子跳跃的检测接着,调查了连接捕获法是否也能够检测报告为在肺腺癌中为致癌性的met外显子14跳跃等的转录产物。从通过使用新鲜的冷冻样品的rna-seq而确定了具有met外显子14跳跃的肺腺癌的病例的五个ffpe提取出rna。对从外显子13向外显子15的连接、即支持外显子14的跳跃的连接支持读出的数目进行了计数。在连接捕获法中,在具有外显子跳跃的所有五个ffpe样品中都鉴定到了met外显子14跳跃,但在不具有met外显子跳跃的其他34的病例中,所有都未看到连接支持读出(图3、表3)。其表示连接捕获法也能够检测外显子跳跃。[表3]实施例3:针对活检样品的连接捕获法的应用此外,评价了连接捕获法是否能够应用于较小的活检样品。从以针活检(coreneedlebipsy)、细针抽吸活检(fineneedleaspiration)以及经支气管镜肺活检(transbronchiallungbiopsy、tblb)为代表的融合基因阳性的ffpe标本制备了rna。令人惊讶的是,在所有rna-seq中,在各标本检测到支持特异的正确融合转录产物的多个连接支持读出(图4、表4)。[表4]实施例4:连接捕获法的临床上的有用性对于通过作为kras以及egfr变异阴性的、阶段ii或iii的nsclc的40病例的外科切除而获得到的ffpe,通过连接捕获法进行实验,从而评价了该方法的临床上的有用性。met外显子14跳跃、eml4-alk融合基因、ret融合基因分在3病例、2病例以及1病例中检测到(未示出数据)。此外,为了评价连接捕获法对肉瘤的诊断的临床上的有用性,在前瞻研究中,对肉瘤患者实施了连接捕获法。在以下的表5中示出结果。[表5]由于在粘液基质附近的具有非典型核的纺锤细胞增殖,因此一个病例(#44)被诊断为了粘液纤维肉瘤。然而,由于通过连接捕获法检测到对血管纤维瘤作为特异性融合基因的ahrr-nco2a基因,从而明确了本病例是软组织血管纤维瘤。另一病例(#48)为taf15-nr4a3阳性,其与骨外软骨肉瘤的诊断结果一致。这些结果表示连接捕获法可使用于疾病的诊断。实施例5:基因表达量的测量在本实施例中,使用连接捕获法进行了基因表达量的测量。(材料和方法)基因表达量测量对于十一种类的持家基因(actb、b2m、gapdh、gusb、h3f3a、hprt1、hsp90ab1、ppia、rplp0、tfrc以及ubc),按照实施例1从ffpe样品提取所有rna,并按照实施例1进行了基于cdna捕获(连接捕获)的rna-seq。为了比较,也按照实施例1从新鲜冷冻样品提取所有rna,并进行了基于多聚a选择的rna-seq。只是,在本实施例中,除了由实施例1所示的用于连接捕获法的客制化探针(toprnav3)之外,还加上基因表达量测量用的探针进行了浓缩。作为基因表达量测量用探针,使用了对包含erbb2等的癌症基因的125基因以2×覆瓦设计的探针。所有探针长都设为120碱基。基于覆瓦数的读出数的校正正如在实施例中记载的那样,为了通过连接捕获法获得尽量多的种类的读出,而在假想连接点附近集中地以5×或10×覆瓦进行了探针的设计。因此,在基于读出数推定基因的表达量的情况下,根据探针的数目而存在较多地算出表达量的可能。因此,在连接捕获法中,通过用读出数除以探针的覆瓦数,从而校正了读出数(例如,如果是5×覆瓦,则用读出数除以5,如果是10×覆瓦,则用读出数除以10)。基于持家基因的读出数的校正由于在连接捕获法中使用了ffpe样品(a组),并在基于多聚a选择的rna-seq中使用了新鲜冷冻样品(b组),因此以使两者的持家基因的表达量变为相等的方式对样品间的质量的差异进行了校正。具体而言,对以使十一种持家基因表达量的a群组和b群组之比的log_2平均为相等的方式校正b群组表达量的系数进行计算,并使用这些系数校正了所有基因的表达量。(结果)对于来源于肺癌患者的七个样品,使用基于多聚a选择的rna-seq以及连接捕获法,对十一种类的持家基因(actb、b2m、gapdh、gusb、h3f3a、hprt1、hsp90ab1、npm1、ppia、rplp0、tfrc以及ubc)的表达量进行了测量。其结果,对于持家基因,在基于多聚a选择的rna-seq和连接捕获法中,rpkm(readsperkilobaseofexonmodelpermillionmappedreads)的值确认到相关性(未示出数据)。接着,对于rna-seq的rpkm和基于连接捕获法中的覆瓦数进行了校正的rpkm,对于表达量测量用基因群组和融合基因解析用基因群组计算出相关系数。在此,表达量测量用基因群组是通过基因表达量测量用的探针进行了表达测量的基因群组,融合基因解析用基因群组是通过用于连接捕获法的客制化探针进行了表达测量的基因群组。在图5a以及表6中示出表达量测量用基因群组的结果,在图5b以及表7中示出融合基因解析用基因群组的结果。在表达量测量用基因群组和融合基因解析用基因群组中,看到rna-seq的rpkm与连接捕获法的rpkm的相关性,特别是对于表达量测量用基因群组看到更强的相关性。其结果表示出,基因表达量测量用的探针适于表达量的测量,但用于连接捕获法的客制化探针也可使用于表达量的测量。此外,其结果表示出,在除了基因表达量测量用的探针之外还包含用于连接捕获法的客制化探针的情况下,也能够准确地测量基因表达量。[表6]样品相关系数样品-10.938599样品-20.971988样品-30.962161样品-40.953048样品-50.991559样品-60.990007样品-70.99219[表7]样品相关系数样品-10.817235样品-20.770109样品-30.860437样品-40.782432样品-50.822337样品-60.630832样品-70.801661实施例6:基于基于表达量的癌症的聚类对来源于luad(肺腺癌)、sarc(肉瘤)、muca(多发癌症)以及lusc(肺扁平上皮癌)的患者的样品,按照实施例5,也加上基因表达量测量用的探针,通过连接捕获法进行了基因表达测量。具体而言,对于表达量测量用、融合基因解析用两者的基因的合计467基因,按照实施例5所记载的方法进行基于覆瓦数的读出数的校正以及基于持家基因的读出数的校正,求出表达值。将所求出的表达值(xn,n=1、……、n,n是基因数)进行对数转换(log_2(xn+1)),基于该值,使用统计解析用软件r的heatmap.3进行了聚类。其结果,如图6所示,基于基因的表达量对luad、sarc、muca以及lusc进行了聚类。其表示通过本发明的方法测量基因表达量而可确定原发癌症的种类。产业实用性本发明提供一种能够简便地检测由融合基因和/或外显子跳跃产生的转录产物的方法。由此,由于能够根据疾病的诊断、受试者的遗传上的背景来选择合适的药剂,因此产业实用性较大。在本说明书中引用的所有刊物、专利以及专利申请,都是直接通过引用而编入本书明书中。序列表<110>国立大学法人东京大学<120>检测由融合基因和/或外显子跳跃产生的转录产物的探针以及方法<130>ph-7477-pct<150>jp2017-125074<151>2017-06-27<160>12<170>patentinversion3.5<210>1<211>136<212>dna<213>人<400>1aaatatgaaaagccaaaatttgtgcagtgtttagcattcttggggaatggagatgttctt60actggagactcaggtggagtcatgcttatatggagcaaaactactgtagagcccacacct120gggaaaggacctaaag136<210>2<211>120<212>dna<213>人工<220><223>探针<400>2aatttgtgcagtgtttagcattcttggggaatggagatgttcttactggagactcaggtg60gagtcatgcttatatggagcaaaactactgtagagcccacacctgggaaaggacctaaag120<210>3<211>120<212>dna<213>人工<220><223>探针<400>3aatatgaaaagccaaaatttgtgcagtgtttagcattcttggggaatggagatgttctta60ctggagactcaggtggagtcatgcttatatggagcaaaactactgtagagcccacacctg120<210>4<211>120<212>dna<213>人工<220><223>探针<400>4agggaatttttgggaaatatgaaaagccaaaatttgtgcagtgtttagcattcttgggga60atggagatgttcttactggagactcaggtggagtcatgcttatatggagcaaaactactg120<210>5<211>120<212>dna<213>人工<220><223>探针<400>5cactaacaagaaaacagggaatttttgggaaatatgaaaagccaaaatttgtgcagtgtt60tagcattcttggggaatggagatgttcttactggagactcaggtggagtcatgcttatat120<210>6<211>120<212>dna<213>人工<220><223>探针<400>6cctggagcggcaattcactaacaagaaaacagggaatttttgggaaatatgaaaagccaa60aatttgtgcagtgtttagcattcttggggaatggagatgttcttactggagactcaggtg120<210>7<211>187<212>dna<213>人<400>7tgtaccgccggaagcaccaggagctgcaagccatgcagatggagctgcagagccctgagt60acaagctgagcaagctccgcacctcgaccatcatgaccgactacaaccccaactactgct120ttgctggcaagacctcctccatcagtgacctgaaggaggtgccgcggaaaaacatcaccc180tcattcg187<210>8<211>120<212>dna<213>人工<220><223>探针<400>8tgtaccgccggaagcaccaggagctgcaagccatgcagatggagctgcagagccctgagt60acaagctgagcaagctccgcacctcgaccatcatgaccgactacaaccccaactactgct120<210>9<211>120<212>dna<213>人工<220><223>探针<400>9caccaggagctgcaagccatgcagatggagctgcagagccctgagtacaagctgagcaag60ctccgcacctcgaccatcatgaccgactacaaccccaactactgctttgctggcaagacc120<210>10<211>120<212>dna<213>人工<220><223>探针<400>10gccatgcagatggagctgcagagccctgagtacaagctgagcaagctccgcacctcgacc60atcatgaccgactacaaccccaactactgctttgctggcaagacctcctccatcagtgac120<210>11<211>120<212>dna<213>人工<220><223>探针<400>11ctgcagagccctgagtacaagctgagcaagctccgcacctcgaccatcatgaccgactac60aaccccaactactgctttgctggcaagacctcctccatcagtgacctgaaggaggtgccg120<210>12<211>120<212>dna<213>人工<220><223>探针<400>12gtacaagctgagcaagctccgcacctcgaccatcatgaccgactacaaccccaactactg60ctttgctggcaagacctcctccatcagtgacctgaaggaggtgccgcggaaaaacatcac120当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1