热激活延迟荧光化合物、发光材料及有机电致发光器件的制作方法
本发明涉及热激活延迟荧光化合物、包含该热激活延迟荧光化合物的发光材料以及包含该热激活延迟荧光化合物的有机电致发光器件。
背景技术:
:有机电致发光器件(oled)具有自主发光、低电压驱动、全固化、宽视角、组成和工艺简单等一系列的优点,与液晶显示器相比,oled不需要背光源。因此,oled具有广泛的应用前景。众所周知,在电场激发下,激子一般由25%单重态和75%三重态激子构成。然而,三重态激子的寿命较长,易通过非辐射的方式耗散。因此,如何利用三重态激子来发光是oled领域的关键问题。基于铱、铂等磷光材料的第二代oled,通过重原子效应产生强烈的自旋轨道耦合(soc)作用,增强了单重态到三重态系间窜越(isc),使器件的内部量子效率(iqe)达到100%。但因材料需采用稀有金属,成本较高,给磷光材料的广泛应用和全面商业化增加了难度。为了解决这一问题,2009年日本九州大学adachi教授提出了不依赖于铱、铂等贵金属,其iqe也能达到100%的第三代纯有机电致发光材料设计理论(nature,2012,492,234-238)。这种所谓的第三代有机电致发光材料,一般具有小的单线态-三线态能级差(δest),三线态激子可以通过反向系间窜越(risc)转变成单线态激子发光,这可以同时利用电激发下形成的单重态激子和三重态激子,器件的内部量子效率可达100%,因此被视为未来广泛应用的有机发光材料之一。然而,在这一领域中,器件的工作寿命,特别是蓝光器件的工作寿命,仍然是一个悬而未决的问题。因此,现有技术还有待改进和发展。技术实现要素:发明要解决的问题根据本发明,提供一种热激活延迟荧光化合物,其可用作发光材料的构成材料,且可用于有机电致发光器件发光层的发光客体。本发明的热激活延迟荧光化合物可以克服现有技术的材料的缺陷,诸如低效率、短寿命以及高驱动电压等问题。本发明的目的在于,提供一种能有效提高分子的玻璃化转变温度、荧光量子产率和载流子迁移率的热激活延迟荧光化合物。本发明的另一个目的是提供一种包含该热激活延迟荧光化合物的发光材料,该发光材料能够作为高效率的有机电致发光器件用的材料,特别是作为发光层的荧光客体材料。本发明的又一目的是提供一种包含该热激活延迟荧光化合物的有机电致发光器件,本发明的有机电致发光器件具有低驱动电压、长器件寿命和高发光效率。用于解决问题的方案令人惊讶地发现,下面描述的化合物能有效提高分子的玻璃化转变温度、荧光量子产率和载流子迁移率,进而将其用于有机电致发光器件时,能够有效降低驱动电压,并且提高器件寿命和效率。即,本发明如下。[1]一种热激活延迟荧光化合物,其由下述通式(1)表示:其中,x表示0~4的整数,y和z各自独立地表示0~6的整数,且x+y+z=6;a表示吸电子基团,d1和d2各自独立地表示给电子基团或氢原子。[2]根据[1]所述的热激活延迟荧光化合物,其中,前述吸电子基团表示三氟甲基、氰基、取代或未取代的酰基基团、取代或未取代的嘧啶基团、取代或未取代的均三嗪基团、取代或未取代的二苯甲酮基团、取代或未取代的二苯砜基团、取代或未取代的1-苯基-1h-苯并咪唑基团、取代或未取代的蒽醌基团、取代或未取代的噻蒽-5,5,10,10-四氧化物基团、取代或未取代的硫杂蒽酮基团、取代或未取代的硫代呫吨-9-酮基团、取代或未取代的硫代呫吨-9-酮-10,10-二氧化物基团、或取代或未取代的间苯二腈基团。[3]根据[1]所述的热激活延迟荧光化合物,其中,前述吸电子基团选自下述基团:[4]根据[1]所述的热激活延迟荧光化合物,其中,前述给电子基团为取代或未取代的咔唑基团、取代或未取代的苯并噻吩并咔唑基团、取代或未取代的苯并呋喃并咔唑基团、取代或未取代的吲哚并咔唑基团、取代或未取代的苯并咪唑并苯并咪唑基团、取代或未取代的茚并[1,2-c]咔唑基团、或取代或未取代的亚氨基二苄基团。[5]根据[1]所述的热激活延迟荧光化合物,其中,前述给电子基团选自下述基团:其中,r1、r2、r3和r4各自独立地表示氢原子、氘原子、卤素原子、氰基、具有1至40个碳原子的烷基、具有1至40个碳原子的烷氧基、具有1至40个碳原子的硫代烷基基团、具有6至30个碳原子的芳香族烃基、或具有5至30个碳原子的芳香族杂环基。[6]根据[1]~[5]中任一项所述的热激活延迟荧光化合物,其中,由前述通式(1)表示的化合物选自下述化合物:[7]一种发光材料,其包含[1]~[6]中任一项所述的热激活延迟荧光化合物。[8]一种有机电致发光器件,其包括:第一电极、与第一电极对置而具备的第二电极、以及夹在第一电极与第二电极之间的至少一个有机层,其中有机层包含[1]~[6]中任一项所述的热激活延迟荧光化合物。[9]根据[8]所述的有机电致发光器件,其中,包含所述热激活延迟荧光化合物的有机层为发光层。发明的效果本发明的热激活延迟荧光化合物能够有效提高分子的玻璃化转变温度、荧光量子产率、载流子迁移率。另外,本发明的热激活延迟荧光发光材料原料的合成更简单,成本更低。此外,本发明的热激活延迟荧光化合物作为发光材料的构成材料是有用的。本发明的发光材料能够得到以下效果:(1)本发明提供一种全新的热激活延迟荧光发光材料,其单线态-三线态能隙(<0.2ev),可有效地发出延迟荧光。(2)本发明的热激活延迟荧光发光材料具有较高的玻璃化转变温度(>100℃),保证了其在器件制备过程中具有较好的成膜性,从而可以提高器件寿命。(3)本发明的热激活延迟荧光发光材料具有较高的荧光量子效率(>80%),这主要得益于所设计分子具有一定的分子刚性,大大减弱了其激发态到基态的非辐射跃迁。另外,本发明的热激活延迟荧光化合物作为有机电致发光器件的发光层的荧光客体材料是有用的,通过使用该化合物制作有机电致发光器件,可以得到高发光效率、低驱动电压和长器件寿命的有机电致发光器件。附图说明图1为本发明实施例4的化合物(化合物1-22)的紫外吸收光谱(uv-vis)、室温荧光光谱(pl)和低温磷光光谱(phos);紫外吸收光谱、室温荧光光谱和低温磷光光谱在甲苯的稀溶液(1×10-5mol/l)中测得。图2为本发明实施例7、9和10中有机电致发光器件1、3和4的有机电致发光光谱。图3是显示实施例7~10的有机电致发光器件和比较例1的有机电致发光器件的构造的图。附图标记说明1基板2阳极3空穴注入层4空穴传输层5电子阻挡层6发光层7空穴阻挡层8电子传输层9电子注入层10阴极具体实施方式以下,对本发明的实施方式进行详细说明。但是,本发明不受以下实施方式的限定。本发明的热激活延迟荧光化合物由下述通式(1)表示。在通式(1)中,x表示0~4的整数,y和z各自独立地表示0~6的整数,且x+y+z=6;a表示吸电子基团,d1和d2各自独立地表示给电子基团或氢原子。本发明的热激活延迟荧光化合物中,给电子基团和吸电子基团因为大位阻的原因具有较大扭曲角,分子的最高占据分子轨道(homo)和最低未占分子轨道(lumo)能够有效的分离,分别分布在给电子基团和吸电子基团上。homo与lumo的空间分离能够减小单线态和三线态能隙,从而实现其能隙差小于0.2ev。材料的能隙可以通过室温荧光光谱和低温磷光光谱得到。在优选的实施方式中,前述吸电子基团表示三氟甲基、氰基、取代或未取代的酰基基团、取代或未取代的嘧啶基团、取代或未取代的均三嗪基团、取代或未取代的二苯甲酮基团、取代或未取代的二苯砜基团、取代或未取代的1-苯基-1h-苯并咪唑基团、取代或未取代的蒽醌基团、取代或未取代的噻蒽-5,5,10,10-四氧化物基团、取代或未取代的硫杂蒽酮基团、取代或未取代的硫代呫吨-9-酮基团、取代或未取代的硫代呫吨-9-酮-10,10-二氧化物基团、或取代或未取代的间苯二腈基团。作为前述吸电子基团所示的“取代的嘧啶基团”中的“取代基”,可列举出氘原子、氰基、硝基;氟原子、氯原子、溴原子、碘原子等卤素原子;甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、异戊基、新戊基、正己基等具有1至6个碳原子的直链状或支链状的烷基;环戊基、3-甲基环戊基、2,3-二甲基环戊基、环己基、3-甲基环己基、4-甲基环己基、2,3-二甲基环己基、3,4,5-三甲基环己基、4-叔丁基环己基、环庚基、环辛基等具有5至10个碳原子的环烷基;甲氧基、乙氧基、丙氧基等具有1至6个碳原子的直链状或支链状的烷氧基;乙烯基、烯丙基等烯基;苯氧基、甲苯氧基等芳氧基;苄氧基、苯乙氧基等芳基烷氧基;苯基、联苯基、三联苯基、萘基、蒽基、菲基、芴基、茚基、芘基、苝基、荧蒽基等芳香族烃基;吡啶基、嘧啶基、三嗪基、呋喃基、噻吩基、吡咯基、吡唑基等芳香族杂环基;苯乙烯基、萘基乙烯基等芳基乙烯基;乙酰基、苯甲酰基等酰基之类基团。这些取代基之中,优选苯基。另外,这些取代基之间可以介由单键、取代或未取代的亚甲基、氧原子或硫原子彼此键合而形成环。作为前述吸电子基团所示的“取代的均三嗪基团”中的“取代基”,可列举出与关于前述吸电子基团所示的“取代的嘧啶基团”的“取代基”示出的基团相同的基团,能够采用的方式也可列举出同样的方式。优选地,前述吸电子基团所示的“取代的均三嗪基团”的“取代基”为苯基和环己基。作为前述吸电子基团所示的“取代的酰基基团”、“取代的二苯甲酮基团”、“取代的二苯砜基团”、“取代的1-苯基-1h-苯并咪唑基团”、“取代的蒽醌基团”、“取代的噻蒽-5,5,10,10-四氧化物基团”、“取代的硫杂蒽酮基团”、“取代的硫代呫吨-9-酮基团”、“取代的硫代呫吨-9-酮-10,10-二氧化物基团”和“取代的间苯二腈基团”中的“取代基”,可列举出与关于前述吸电子基团所示的“取代的嘧啶基团”的“取代基”示出的基团相同的基团,能够采用的方式也可列举出同样的方式。前述吸电子基团优选选自下述基团:在优选的实施方式中,前述给电子基团为取代或未取代的咔唑基团、取代或未取代的苯并噻吩并咔唑基团、取代或未取代的苯并呋喃并咔唑基团、取代或未取代的吲哚并咔唑基团、取代或未取代的苯并咪唑并苯并咪唑基团、取代或未取代的茚并[1,2-c]咔唑基团、或取代或未取代的亚氨基二苄基团。作为前述给电子基团所示的“取代的咔唑基团”、“取代的苯并噻吩并咔唑基团”、“取代的苯并呋喃并咔唑基团”、“取代的吲哚并咔唑基团”、“取代的苯并咪唑并苯并咪唑基团”、“取代的茚并[1,2-c]咔唑基团”和“取代的亚氨基二苄基团”中的“取代基”,可列举出氘原子;氰基;氟原子、氯原子、溴原子、碘原子等卤素原子;甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、异戊基、新戊基、正己基、正庚基、2-甲基己基、正辛基、异辛基、叔辛基、2-乙基己基、3-甲基庚基、正壬基、正癸基、十六烷基、十八烷基、二十烷基、环丙基、环丁基、环戊基、3-甲基环戊基、2,3-二甲基环戊基、环己基、3-甲基环己基、4-甲基环己基、2,3-二甲基环己基、3,4,5-三甲基环己基、4-叔丁基环己基、环庚基、环辛基等具有1至40个碳原子的直链、支链或环状烷基;甲氧基、乙氧基、正丙氧基、异丙氧基、异丙基氧基、正丁氧基、异丁氧基、叔丁氧基、仲丁氧基、正戊氧基、新戊氧基、异戊氧基、正己氧基、3,3-二甲基丁氧基、2-乙基丁氧基、正辛氧基、正壬氧基、正癸氧基、苄氧基、对甲基苄氧基等具有1至40个碳原子的直链、支链或环状烷氧基;甲硫基、乙硫基、正丙硫基、异丙硫基、正丁硫基、异丁硫基、仲丁硫基、叔丁硫基、正戊硫基、仲戊硫基、正己硫基、环己硫基、正庚硫基、环庚硫基、正辛硫基、环辛硫基、2-乙基己硫基等具有1至40个碳原子的直链、支链或环状硫代烷基基团;苯基、联苯基、三联苯基、萘基、蒽基、菲基、芴基、茚基、芘基、苝基、荧蒽基、苯并菲基等具有6至30个碳原子的芳香族烃基;吡啶基、嘧啶基、三嗪基、呋喃基、噻吩基、吡咯基、喹啉基、异喹啉基、苯并呋喃基、苯并噻吩基、吲哚基、咔唑基、苯并噁唑基、苯并噻唑基、喹喔啉基、苯并咪唑基、吡唑基、二苯并呋喃基、二苯并噻吩基、咔啉基等具有5至30个碳原子的芳香族杂环基。另外,这些取代基之间可以介由单键、取代或未取代的亚甲基、氧原子或硫原子彼此键合而形成环。在优选的实施方式中,前述给电子基团优选选自下述基团:其中,r1、r2、r3和r4各自独立地表示氢原子、氘原子、卤素原子、氰基、具有1至40个碳原子的烷基、具有1至40个碳原子的烷氧基、具有1至40个碳原子的硫代烷基基团、具有6至30个碳原子的芳香族烃基、或具有5至30个碳原子的芳香族杂环基。作为前述r1~r4所示的“卤素原子”,例如可列举出:氟原子、氯原子、溴原子或碘原子。作为前述r1~r4所示的“具有1至40个碳原子的烷基”,例如可以为直链、支链或环状烷基。具体而言,具有1至40个碳原子的烷基可以为具有1至40个碳原子的直链烷基或具有3至40个碳原子的支链或环状烷基。作为具体实例,例如可列举出:甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、异戊基、新戊基、正己基、正庚基、2-甲基己基、正辛基、异辛基、叔辛基、2-乙基己基、3-甲基庚基、正壬基、正癸基、十六烷基、十八烷基、二十烷基、环丙基、环丁基、环戊基、3-甲基环戊基、2,3-二甲基环戊基、环己基、3-甲基环己基、4-甲基环己基、2,3-二甲基环己基、3,4,5-三甲基环己基、4-叔丁基环己基、环庚基、环辛基等。作为前述r1~r4所示的“具有1至40个碳原子的烷氧基”,例如可以为直链、支链或环状烷氧基。具体而言,具有1至40个碳原子的烷氧基可以为具有1至40个碳原子的直链烷氧基或具有3至40个碳原子的支链或环状烷氧基。作为具体实例,例如可列举出:甲氧基、乙氧基、正丙氧基、异丙氧基、异丙基氧基、正丁氧基、异丁氧基、叔丁氧基、仲丁氧基、正戊氧基、新戊氧基、异戊氧基、正己氧基、3,3-二甲基丁氧基、2-乙基丁氧基、正辛氧基、正壬氧基、正癸氧基、苄氧基、对甲基苄氧基等。作为前述r1~r4所示的“具有1至40个碳原子的硫代烷基基团”,例如可以为直链、支链或环状硫代烷基基团。具体而言,具有1至40个碳原子的硫代烷基基团可以为具有1至40个碳原子的直链硫代烷基基团或具有3至40个碳原子的支链或环状硫代烷基基团。作为具体实例,例如可列举出:甲硫基、乙硫基、正丙硫基、异丙硫基、正丁硫基、异丁硫基、仲丁硫基、叔丁硫基、正戊硫基、仲戊硫基、正己硫基、环己硫基、正庚硫基、环庚硫基、正辛硫基、环辛硫基、2-乙基己硫基等。作为前述r1~r4所示的“具有6至30个碳原子的芳香族烃基”,例如可列举出:苯基、联苯基、三联苯基、萘基、蒽基、菲基、芴基、茚基、芘基、苝基、荧蒽基、苯并菲基等。作为前述r1~r4所示的“具有5至30个碳原子的芳香族杂环基”,例如可列举出:吡啶基、嘧啶基、三嗪基、呋喃基、噻吩基、吡咯基、喹啉基、异喹啉基、苯并呋喃基、苯并噻吩基、吲哚基、咔唑基、苯并噁唑基、苯并噻唑基、喹喔啉基、苯并咪唑基、吡唑基、二苯并呋喃基、二苯并噻吩基、咔啉基等。在本发明的热激活延迟荧光化合物中,优选的化合物的具体实例如下所示,但本发明不限于这些化合物。本发明的热激活延迟荧光化合物的纯化通过利用柱色谱的纯化、利用硅胶、活性炭、活性白土等的吸附纯化、利用溶剂的重结晶、析晶法、升华纯化法等来进行。热激活延迟荧光化合物的鉴定利用质谱、元素分析进行。<发光材料>本发明的发光材料包含上述热激活延迟荧光化合物。本发明的发光材料可用于有机电致发光器件中,尤其作为有机电致发光器件的荧光客体材料。<有机电致发光器件>本发明的有机电致发光器件包括:第一电极、与第一电极对置而具备的第二电极、以及夹在第一电极与第二电极之间的至少一个有机层,其中有机层包含本发明的热激活延迟荧光化合物。图3是显示本发明的有机电致发光器件的构造的图。如图3所示,在本发明的有机电致发光器件中,例如,阳极2、空穴注入层3、空穴传输层4、电子阻挡层5、发光层6、空穴阻挡层7、电子传输层8、电子注入层9和阴极10依次设置在基板1上。本发明的有机电致发光器件不限于这样的结构,例如在该多层结构中,可以省略一些有机层。例如,可以是阳极2与空穴传输层4之间的空穴注入层3、发光层6与电子传输层8之间的空穴阻挡层7、以及电子传输层8与阴极10之间的电子注入层9被省略,并且在基板1上依次设置阳极2、空穴传输层4、发光层6、电子传输层8和阴极10的构型。根据本发明的有机电致发光器件除了上述有机层包含由上述通式(1)表示的化合物以外,可以通过本领域
技术领域:
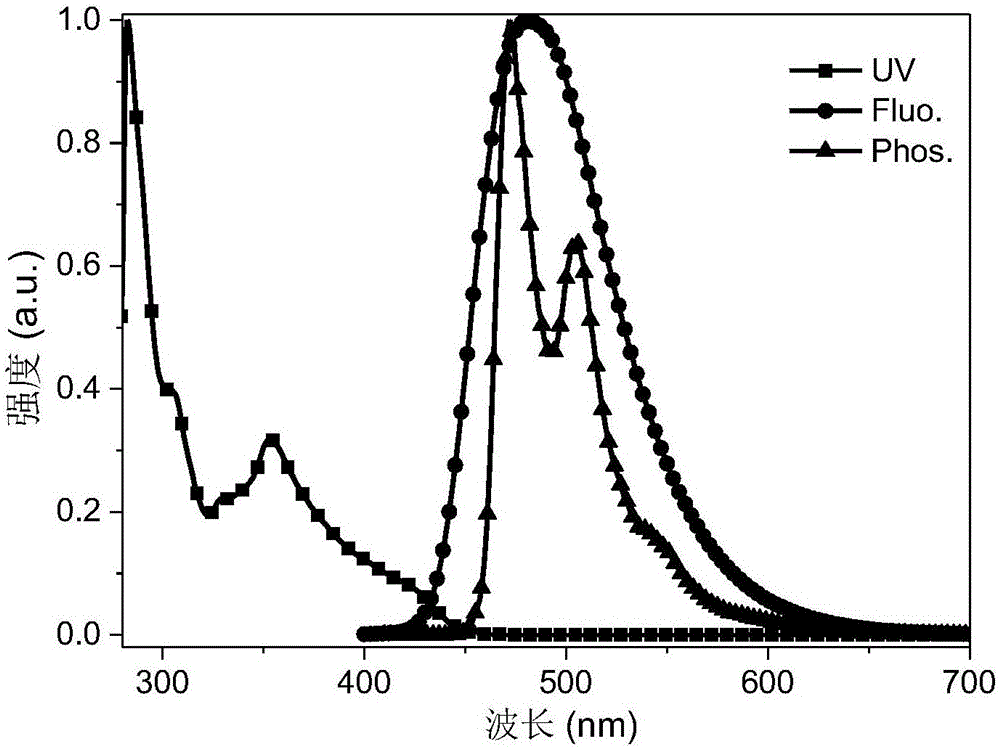
公知的材料和方法进行制造。另外,在上述有机电致发光器件包含多个有机层的情况下,上述有机层可以由相同的物质或不同的物质形成。例如,根据本发明的有机电致发光器件可以通过在基板上依次层叠第一电极、有机层和第二电极而制造。此时,可以如下制造:利用溅射法或电子束蒸发法之类的pvd(物理蒸镀法)方法,在基板上蒸镀金属或具有导电性的金属氧化物或它们的合金而形成阳极,然后在该阳极上形成包含空穴注入层、空穴传输层、发光层和电子传输层的有机层,之后在该有机物层上蒸镀可用作阴极的物质。但是,制造方法并不限定于此。作为一个例子,上述第一电极为阳极,上述第二电极为阴极,或者上述第一电极为阴极,上述第二电极为阳极。作为本发明的有机电致发光器件的阳极,可以由公知的电极材料构成。例如使用具有大的功函数的电极材料,如钒、铬、铜、锌、金等金属或它们的合金;如氧化锌、氧化铟、氧化铟锡(ito)、氧化铟锌(izo)等金属氧化物;如zno:al或sno2:sb等金属与氧化物的组合;聚(3-甲基噻吩)、聚[3,4-(亚乙基-1,2-二氧)噻吩](pedot)、聚吡咯和聚苯胺等导电性高分子等。这些之中,优选ito。作为本发明的有机电致发光器件的空穴注入层,可以使用公知的具有空穴注入性质的材料。例如可列举:以铜酞菁为代表的卟啉化合物、萘二胺衍生物、星型的三苯胺衍生物、分子中具有3个以上三苯胺结构通过单键或不含杂原子的二价基团连接的结构的芳胺化合物等三苯胺三聚体及四聚体、六氰基氮杂苯并菲等受体型杂环化合物、涂布型高分子材料。这些材料可以通过蒸镀法、以及旋涂法、喷墨法等公知方法来形成薄膜。作为本发明的有机电致发光器件的空穴传输层,可以使用公知的具有空穴传输性的材料。例如可列举:含有间咔唑基苯基的化合物;如n,n’-二苯基-n,n’-二(间甲苯基)联苯胺(tpd)、n,n'-二苯基-n,n'-(1-萘基)-1,1'-联苯-4,4'-二胺(npb)、n,n,n’,n’-四联苯基联苯胺等联苯胺衍生物;1,1-双[(二-4-甲苯基氨基)苯基]环己烷(tapc);各种三苯胺三聚体及四聚体;9,9',9”-三苯基-9h,9'h,9”h-3,3':6',3”-三咔唑(tris-pcz)等。它们可以单独地成膜,也可以以与其它材料一起混合成膜而成的单层的形式来使用,还可以制成单独成膜而成的层彼此的层叠结构、混合成膜而成的层彼此的层叠结构、或者单独成膜而成的层与混合成膜而成的层的层叠结构。这些材料可以通过蒸镀法、以及旋涂法、喷墨法等公知方法来形成薄膜。此外,空穴注入层或空穴传输层中,也可以使用对于该层中通常使用的材料进一步p掺杂三溴苯胺六氯化锑、轴烯衍生物等而成的物质、其部分结构中具有tpd等联苯胺衍生物的结构的高分子化合物等。作为本发明的有机电致发光器件的电子阻挡层,可以使用具有电子阻挡作用的公知化合物形成。例如,可列举出:3,3'-二(n-咔唑基)-1,1'-联苯(mcbp)、4,4’,4”-三(n-咔唑基)三苯胺(tcta)、9,9-双[4-(咔唑-9-基)苯基]芴、1,3-双(咔唑-9-基)苯(mcp)、2,2-双(4-咔唑-9-基苯基)金刚烷(ad-cz)等咔唑衍生物;9-[4-(咔唑-9-基)苯基]-9-[4-(三苯基甲硅烷基)苯基]-9h-芴所代表的具有三苯基甲硅烷基和三芳胺结构的化合物;电子阻挡性高的单胺化合物、各种三苯胺二聚体等具备电子阻挡作用的化合物。它们可以单独地成膜,也可以与其它材料一起混合成膜而成的单层的形式来使用,还可以制成单独成膜而成的层彼此的层叠结构、混合成膜而成的层彼此的层叠结构、或者单独成膜而成的层与混合成膜而成的层的层叠结构。这些材料可以通过蒸镀法、以及旋涂法、喷墨法等公知方法来形成薄膜。作为本发明的有机电致发光器件的发光层,优选使用包含通式(1)所示的热激活延迟荧光化合物的发光材料。除此之外,还可以使用以alq3为首的羟基喹啉衍生物的金属络合物等各种金属络合物、具有嘧啶环结构的化合物、蒽衍生物、双苯乙烯基苯衍生物、芘衍生物、噁唑衍生物、聚对亚苯基亚乙烯基衍生物等。发光层可以由主体材料和掺杂材料构成。作为主体材料,例如可以使用mcbp、mcp、噻唑衍生物、苯并咪唑衍生物、聚二烷基芴衍生物、具有吲哚环作为稠合环的部分结构的杂环化合物等。作为掺杂材料,优选使用包含通式(1)所示的热激活延迟荧光化合物的发光材料。本发明的热激活延迟荧光化合物的掺杂重量比例优选为0.1~50wt%,更优选为0.5~20wt%,特别优选为0.5~8wt%。掺杂材料除了使用本发明的热激活延迟荧光化合物之外,还可以使用芘衍生物、蒽衍生物、喹吖啶酮、香豆素、红荧烯、苝和它们的衍生物、苯并吡喃衍生物、罗丹明衍生物、氨基苯乙烯基衍生物、螺环双芴衍生物等。它们可以单独地成膜,也可以与其它材料一起混合成膜而成的单层的形式来使用,还可以制成单独成膜而成的层彼此的层叠结构、混合成膜而成的层彼此的层叠结构、或者单独成膜而成的层与混合成膜而成的层的层叠结构。这些材料可以通过蒸镀法、以及旋涂法、喷墨法等公知方法来形成薄膜。作为本发明的有机电致发光器件的空穴阻挡层,可以使用公知的具有空穴阻挡性的化合物来形成。例如可以使用2,4,6-三(3-苯基)-1,3,5-三嗪(t2t)、1,3,5-三(1-苯基-1h-苯并咪唑-2-基)苯(tpbi)、浴铜灵(bcp)等菲咯啉衍生物、铝(iii)双(2-甲基-8-羟基喹啉)-4-苯基苯酚盐(balq)等喹啉醇衍生物的金属络合物、以及各种稀土类络合物、噁唑衍生物、三唑衍生物、三嗪衍生物等具有空穴阻挡作用的化合物。它们可以单独地成膜,也可以以与其它材料一起混合成膜而成的单层的形式来使用,也可以制成单独成膜而成的层彼此的层叠结构、混合成膜而成的层彼此的层叠结构、或单独成膜而成的层与混合成膜而成的层的层叠结构。这些材料可以通过蒸镀法、以及旋涂法、喷墨法等公知方法来形成薄膜。上述具有空穴阻挡性的材料也可以用于下述的电子传输层的形成。即,通过使用上述公知的具有空穴阻挡性的材料,可以形成同时作为空穴阻挡层和电子传输层的层。作为本发明的有机电致发光器件的电子传输层,使用公知的具有电子传输性的化合物形成。例如可以使用alq3、balq为首的羟基喹啉衍生物的金属络合物;各种金属络合物;三唑衍生物;三嗪衍生物;噁二唑衍生物;吡啶衍生物;双(10-羟基苯并[h]喹啉)铍(be(bq2));如2-[4-(9,10-二萘-2-蒽-2-基)苯基]-1-苯基-1h-苯并咪唑(zadn)等苯并咪唑衍生物;噻二唑衍生物;蒽衍生物;碳二亚胺衍生物;喹喔啉衍生物;吡啶并吲哚衍生物;菲咯啉衍生物;噻咯衍生物等。它们可以单独地成膜,也可以以与其它材料一起混合成膜而成的单层的形式来使用,也可以制成单独成膜而成的层彼此的层叠结构、混合成膜而成的层彼此的层叠结构、或单独成膜而成的层与混合成膜而成的层的层叠结构。这些材料可以通过蒸镀法、以及旋涂法、喷墨法等公知方法来形成薄膜。作为本发明的有机电致发光器件的电子注入层,可以使用本身公知的材料形成。例如可以使用氟化锂、氟化铯等碱金属盐;氟化镁等碱土金属盐;羟基喹啉锂等羟基喹啉衍生物的金属络合物;氧化铝等金属氧化物等。在电子注入层或电子传输层中,对于该层中通常使用的材料,可以使用进而n掺杂铯等金属、三芳基氧化膦衍生物等而成的材料。作为本发明的有机电致发光器件的阴极,优选使用具有低功函数的电极材料如铝、镁,或者具有低功函数的合金如镁银合金、镁铟合金、铝镁合金作为电极材料。作为本发明的基板,可以使用传统的有机发光器件中的基板,例如玻璃或塑料。在本发明中,选用玻璃基板。实施例在以下实施例中对由上述通式(1)表示的化合物及包含其的有机电致发光器件的制造进行具体说明。但是,下述实施例仅用于对本发明进行例示,本发明的范围不限于此。实施例1:化合物1-2的合成化合物1-2的合成路线如下所示:在100mlschlenk瓶中,加入3,4,5-三氟苯腈1.00g(6.37mmol),5h-[1]苯并噻吩并[3,2-c]咔唑3.20g(11.70mmol),碳酸铯7.26g(22.28mmol),n,n-二甲基甲酰胺100ml,在氩气保护下,回流搅拌反应12小时,反应完毕。冷却至室温后,倒入水中,淡黄色固体析出,抽滤得到粗产物。将抽滤得到的粗产物经柱层析分离(硅胶为350目,淋洗液为石油醚:二氯甲烷=2:1(v/v)),蒸去溶剂,干燥后,得到4.30g淡黄色固体,收率为73.6%。ms(ei):m/z917.05[m+].anal.calcdforc61h32n4s3(%):c79.89,h3.52,n6.11,s10.49;found:c79.85,h3.60,n6.08,s10.47。实施例2:化合物1-16的合成化合物1-16的合成路线如下所示:在100mlschlenk瓶中,加入2,3,4,5-四氟苯腈0.50g(2.86mmol),5h-[1]苯并噻吩并[3,2-c]咔唑3.51g(12.85mmol),碳酸铯4.65g(14.28mmol),n,n-二甲基甲酰胺60ml,在氩气保护下,回流搅拌反应12小时,反应完毕。冷却至室温后,倒入水中,黄色固体析出,抽滤得到粗产物。将抽滤得到的粗产物经柱层析分离(硅胶为350目,淋洗液为石油醚:二氯甲烷=2:1(v/v)),蒸去溶剂,干燥后,得到2.75g黄色固体,收率为81.1%。ms(ei):m/z1187.54[m+].anal.calcdforc79h41n5s4(%):c79.84,h3.48,n5.89,s10.79;found:c79.79,h3.54,n5.82,s10.85。实施例3:化合物1-21的合成化合物1-21的合成路线如下所示:化合物1-21a的合成在100mlschlenk瓶中,加入2,3,4,5,6-五氟苯腈1.00g(5.18mmol),5h-[1]苯并噻吩并[3,2-c]咔唑4.25g(15.54mmol),碳酸铯5.91g(18.13mmol),n,n-二甲基甲酰胺60ml,在氩气保护下,回流搅拌反应12小时,反应完毕。冷却至室温后,倒入水中,黄色固体析出,抽滤得到粗产物。将抽滤得到的粗产物经柱层析分离(硅胶为350目,淋洗液为石油醚:二氯甲烷=2:1(v/v)),蒸去溶剂,干燥后,得到3.65g黄色固体,收率为73.9%。ms(ei):m/z952.33[m+].anal.calcdforc61h30f2n4s3(%):c76.87,h3.17,f3.99,n5.88,s10.09;found:c76.83,h3.14,f4.03,n5.80,s10.20。化合物1-21的合成在100mlschlenk瓶中,加入化合物1-21a3.00g(3.15mmol),咔唑1.58g(9.44mmol),碳酸铯3.59g(11.02mmol),n,n-二甲基甲酰胺60ml,在氩气保护下,回流搅拌反应12小时,反应完毕。冷却至室温后,倒入水中,黄色固体析出,抽滤得到粗产物。将抽滤得到的粗产物经柱层析分离(硅胶为350目,淋洗液为石油醚:二氯甲烷=2:1(v/v)),蒸去溶剂,干燥后,得到3.05g黄色固体,收率为77.7%。ms(ei):m/z1246.37[m+].anal.calcdforc85h46n6s3(%):c81.84,h3.72,n6.74,s7.71;found:c81.78,h3.78,n6.70,s7.74。实施例4:化合物1-22的合成化合物1-22的合成路线如下所示:化合物1-22a的合成在250mlschlenk瓶中,加入2,3,4,5,6-五氟苯腈4.00g(20.72mmol),咔唑10.39g(62.15mmol),碳酸铯23.63g(72.51mmol),n,n-二甲基甲酰胺150ml,在氩气保护下,回流搅拌反应12小时,反应完毕。冷却至室温后,倒入水中,黄色固体析出,抽滤得到粗产物。将抽滤得到的粗产物经柱层析分离(硅胶为350目,淋洗液为石油醚:二氯甲烷=2.5:1(v/v)),蒸去溶剂,干燥后,得到10.15g黄色固体,收率为77.19%。ms(ei):m/z634.56[m+].anal.calcdforc43h24f2n4(%):c81.37,h3.81,f5.99,n8.83;found:c81.30,h3.84,f5.94,n8.92。化合物1-22的合成在250mlschlenk瓶中,加入1-22a4.00g(6.30mmol),5h-[1]苯并噻吩并[3,2-c]咔唑3.79g(13.87mmol),碳酸铯6.16g(18.91mmol),n,n-二甲基甲酰胺150ml,在氩气保护下,回流搅拌反应12小时,反应完毕。冷却至室温后,倒入水中,黄色固体析出,抽滤得到粗产物。将抽滤得到的粗产物经柱层析分离(硅胶为350目,淋洗液为石油醚:二氯甲烷=2:1(v/v)),蒸去溶剂,干燥后,得到5.70g黄色固体,收率为79.24%。ms(ei):m/z1140.12[m+].anal.calcdforc79h44n6s2(%):c83.13,h3.89,n7.36,s5.62;found:c83.18,h3.88,n7.30,s5.64。实施例5:化合物1-31的合成化合物1-31的合成路线如下所示:在250mlschlenk瓶中,加入3,4,5-三氟苯甲酮2.00g(8.47mmol),3,6-二苯基咔唑8.65g(27.10mmol),碳酸铯9.66g(29.64mmol),n,n-二甲基甲酰胺150ml,在氩气保护下,回流搅拌反应12小时,反应完毕。冷却至室温后,倒入水中,黄色固体析出,抽滤得到粗产物。将抽滤得到的粗产物经柱层析分离(硅胶为350目,淋洗液为石油醚:二氯甲烷=2.5:1(v/v)),蒸去溶剂,干燥后,得到8.10g黄色固体,收率为84.33%。ms(ei):m/z1133.27[m+].anal.calcdforc85h55n3o(%):c90.00,h4.89,n3.70,o1.41;found:c90.18,h4.80,n3.68,o1.34。实施例6:化合物1-33的合成化合物1-33的合成路线如下所示:在250mlschlenk瓶中,加入3,4,5-三氟苯甲酮2.00g(8.47mmol),5h-[1]苯并噻吩并[3,2-c]咔唑7.41g(27.10mmol),碳酸铯9.66g(29.64mmol),n,n-二甲基甲酰胺150ml,在氩气保护下,回流搅拌反应12小时,反应完毕。冷却至室温后,倒入水中,黄色固体析出,抽滤得到粗产物。将抽滤得到的粗产物经柱层析分离(硅胶为350目,淋洗液为石油醚:二氯甲烷=2:1(v/v)),蒸去溶剂,干燥后,得到8.10g黄色固体,收率为79.42%。ms(ei):m/z1133.27[m+].anal.calcdforc67h37n3os3(%):c80.78,h3.74,n4.22,o1.61,s9.65;found:c80.72,h3.85,n4.13,o1.58,s9.72。<玻璃化转变温度的测量>通过高灵敏度差示扫描量热仪(dsc7000x,日立仪器有限公司)测量实施例1~6中得到的化合物的玻璃化转变温度,结果示于表1中。表1玻璃化转变温度(℃)实施例1的化合物(化合物1-2)210实施例2的化合物(化合物1-16)256实施例3的化合物(化合物1-21)275实施例4的化合物(化合物1-22)235实施例5的化合物(化合物1-31)224实施例6的化合物(化合物1-33)186<绝对荧光量子产率的测量>在玻璃基板上,将实施例1的化合物(化合物1-2)和下述结构式的3,3'-二(n-咔唑基)-1,1'-联苯(mcbp)以mcbp的蒸镀速率为2.0nm/s、实施例1的化合物的蒸镀速率为0.16nm/s的蒸镀速率进行双源共蒸,制作膜厚60nm的薄膜,形成有机电致发光器件。通过绝对荧光量子产率测定装置(c9920-02,滨松公司)在室温氮气环境下测定,荧光量子产率示于表2中。除了使用实施例2~6的各化合物代替实施例1的化合物以外,在与实施例1的化合物的绝对荧光量子产率测量相同的条件下测定实施例2~6的化合物的绝对荧光量子产率,结果示于表2中。表2绝对荧光量子产率(%)实施例1的化合物(化合物1-2)86实施例2的化合物(化合物1-16)92实施例3的化合物(化合物1-21)90实施例4的化合物(化合物1-22)88实施例5的化合物(化合物1-31)96实施例6的化合物(化合物1-33)82由表1和表2所示,本发明的化合物发射光谱在蓝色区域,并且具有较高的荧光量子产率和玻璃化转变温度,因此本发明的化合物应用在有机电致发光器件中具有高效率,长寿命的潜力。另外,本发明的化合物具有150℃以上的高玻璃化转变温度,表明本发明的化合物在薄膜状态下是稳定的。实施例7:有机电致发光器件1的制备将空穴注入层3、空穴传输层4、电子阻挡层5、发光层6、空穴阻挡层7、电子传输层8、电子注入层9和阴极10依次形成在预先形成为玻璃基板1上的透明阳极2上,以制备如图3所示的有机电致发光器件。具体地,将形成有膜厚100nm的ito膜的玻璃基板在decon90碱性清洗液中超声处理,去离子水中冲洗,在丙酮和乙醇中各清洗三次,在洁净的环境下烘烤至完全出去水分,用紫外光和臭氧清洗,并用低能阳离子束轰击表面。将该带有ito电极的玻璃基板置入真空腔内,抽真空至2×10-4-2×10-5pa。然后在上述带有ito电极的玻璃基板上以0.2nm/s的蒸镀速率蒸镀2,3,6,7,10,11-六氰基-1,4,5,8,9,12-六氮杂苯并菲(hat-cn)以形成膜厚为10nm的层作为空穴注入层。在空穴注入层上,以2.0nm/s的蒸镀速率蒸镀n,n'-二苯基-n,n'-(1-萘基)-1,1'-联苯-4,4'-二胺(npb)以形成膜厚为40nm的层作为空穴传输层。在空穴传输层上,以2.0nm/s的蒸镀速率蒸镀3,3'-二(n-咔唑基)-1,1'-联苯(mcbp)以形成膜厚为15nm的层作为电子阻挡层。在电子阻挡层上,以3,3'-二(n-咔唑基)-1,1'-联苯(mcbp)的蒸镀速率为2.0nm/s、实施例2的化合物(化合物1-16)的蒸镀速率为0.16nm/s的蒸镀速率进行双源共蒸,形成膜厚为20nm的层作为发光层,实施例2的化合物(化合物1-16)的掺杂重量比例为8wt%。在发光层上,以2.0nm/s的蒸镀速率蒸镀2,4,6-三(3-苯基)-1,3,5-三嗪(t2t)以形成膜厚为10nm的层作为空穴阻挡层。在空穴阻挡层上,以2.0nm/s的蒸镀速率蒸镀2-[4-(9,10-二萘-2-蒽-2-基)苯基]-1-苯基-1h-苯并咪唑(zadn)以形成膜厚为40nm的层作为电子传输层。在电子传输层上,以0.2nm/s的蒸镀速率蒸镀8-羟基喹啉-锂(liq)以形成膜厚为2nm的层作为电子注入层。最后,以3.0nm/s以上的蒸镀速率蒸镀铝,形成膜厚为100nm的阴极。实施例8~10:有机电致发光器件2~4的制备除了使用实施例3、4和6的化合物(化合物1-21、化合物1-22和化合物1-33)代替实施例2的化合物(化合物1-16)作为发光层的材料以外,在以与有机电致发光器件1相同的条件下制作有机电致发光器件,如表3所示。比较例1:有机电致发光器件比较例的制备除了使用蓝色热激活延迟荧光发光材料双[4-(9,9-二甲基-9,10-二氢吖啶)苯基]硫砜(dmac-dps,5%掺杂)代替实施例2的化合物(化合物1-16)作为发光层的材料以外,在以与有机电致发光器件1相同的条件下制作有机电致发光器件,如表3所示。实施例和比较例涉及的化合物结构如下:在常温下大气中当施加直流电压时,测量实施例7~10中制作的有机电致发光器件1~4和比较例1中制作的有机电子放器件比较例的发光特性。测量结果示于表3中。器件的电流-亮度-电压特性是由带有校正过的硅光电二极管的keithley源测量系(keithley2400sourcemeter、keithley2000currentmeter)完成的,电致发光光谱是由photoresearch公司制造的pr655光谱仪测量的,器件的外部量子效率通过文献adv.mater.,2003,15,1043–1048的方法计算可得。器件寿命的测定为:将发光开始时的发光亮度(初始亮度)设定为1000cd/m2,进行恒定电流驱动直到发光亮度衰减到900cd/m2(对应于90%,其中初始亮度取作100%:90%衰减)的时间。所有器件均在氮气环境中封装。表3由表3可见,与比较例1相比,使用本发明的热激活延迟荧光化合物制备的蓝色电致发光器件的发光效率和器件寿命明显优于采用现有技术中的天蓝色材料dmac-dps所制备的有机电致发光器件。由此可见,与通常的发光材料相比,使用了本发明的热激活延迟荧光化合物的有机电致发光器件能够实现外部量子效率的提高,并且驱动电压低、发光亮度也优异。产业上的可利用性本发明的热激活延迟荧光化合物具有极好的发光效率,优异的材料色纯度和寿命特性。因此,由本发明的热激活延迟荧光化合物可以制备具有极佳使用寿命的有机电致发光器件。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1