吡啶酮衍生物的晶型及制备方法和应用与流程
本发明属于医药化学领域,具体涉及具有流感病毒抑制作用的吡啶酮衍生物的晶型以及它的制备方法和应用。
背景技术:
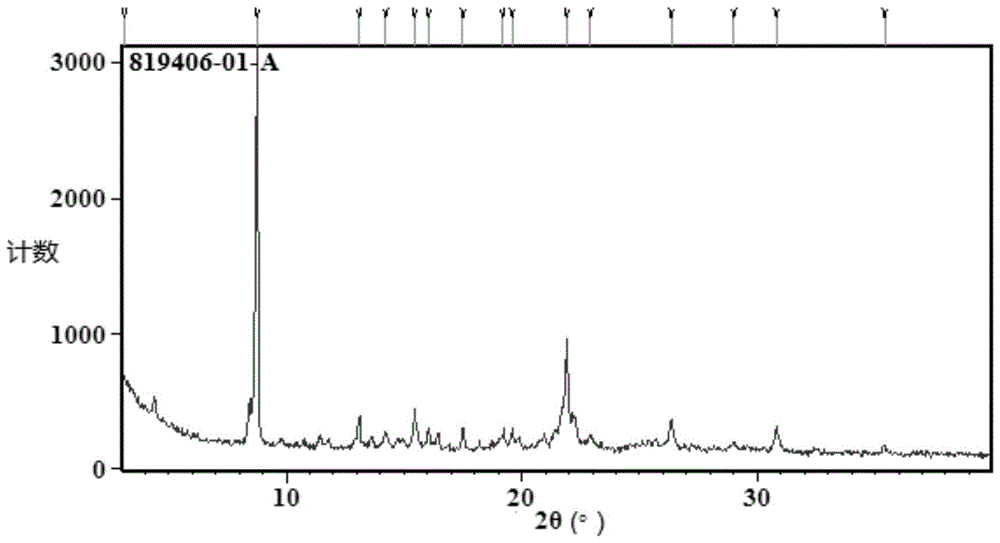
:流感病毒传染性强,容易引起急性呼吸道感染,每年影响5%-20%的成人和20%-30%的儿童。在特殊群体中,如65岁以上老人或者患有慢性基础疾病者,流感病毒容易引起严重的呼吸系统或心血管系统等并发症,在世界范围内每年导致数十万人死亡。神经氨酸酶抑制剂奥司他韦和扎那米韦可以抑制病毒的传播,但是必须在感染后48小时内服用。另外流感病毒容易发生抗原变异,例如1918年的西班牙流感由h1n1亚型引起,1957年的亚洲流感由h2n2亚型引起,1968年的香港流感由h3n2亚型引起,2007年到2008年的流感由h5n1亚型引起。对于新型流感病毒大流行和现有药物产生耐药性的担心,临床上迫切需要全新机理的抗流感药物。流感病毒是rna病毒,其遗传物质是单股负链rna。在流感病毒的生命周期中,病毒反义rna的转录和复制是一个重要的步骤。rna聚合酶由pa、pb1和pb2三个亚基组成,在感染的细胞核内负责病毒rna的转录和复制。流感病毒rna的转录具有特殊的“夺帽”机制,pb2亚基负责识别和结合宿主细胞mrna前体的“帽子结构”,pa亚基负责宿主细胞mrna前体的剪辑,形成引物,启动转录过程。pb1亚基负责病毒mrna的合成。其中pa亚基的帽依赖性内切酶作用,是病毒生命过程所必须的,且具有宿主细胞所不具备的特异性,适合作为全新的靶位开发新型的抗流感药物。此外,药物活性成分的晶型结构往往会引起该药物各种理化性质的差异,如溶解度、溶出速率、熔点、密度、硬度等,这些差异直接影响药物的处方制剂工艺、储存方法、体内药代动力学表现,进而影响到药物的生物利用度、临床疗效和安全性。因此深入研究药物的多晶型现象并找到具备良好性质的晶型具有十分重要的意义。技术实现要素:本申请所要解决的问题是:提供一种新型吡啶酮衍生物,该化合物生物利用度高,在人体服用后可转化为对流感病毒a型和流感病毒b型有极强的抑制活性的化合物,可以单独用于临床治疗或和其他抗流感药物例如神经氨酸酶抑制剂、核苷类药物、pb2抑制剂联合用药,在临床上可快速治愈流感病人。本发明同时还提供了新型吡啶酮衍生物或其溶剂合物的多种结晶,该些结晶在稳定性、引湿性、溶解性等方面符合药用的要求。本发明同时还提供了上述多种结晶的制备方法以及它们在制备抗流感病毒药物中的应用。为解决上述问题,本发明提供的一种技术方案如下:一种式(1)化合物或其溶剂合物的结晶:式(1)化合物的中文名称为:(((r)-12'-((s)-7,8-二氟-6,11-二氢二苯并[b,e]噻庚英-11-基)-6',8'-二氧代-6',8',12',12a'四氢-1'h-,4'h-螺[环丙烷-1,3'-[1,4]恶嗪并[3,4-c]吡啶并[2,1-f][1,2,4]三嗪]-7'-基)氧基)甲基碳酸甲酯;式(1)化合物的英文名称为:(((r)-12'-((s)-7,8-difluoro-6,11-dihydrodibenzo[b,e]thiepin-11-yl)-6',8'-dioxo-6',8',12',12a'-tetrahydro-1'h,4'h-spiro[cyclopropane-1,3'-[1,4]oxazino[3,4-c]pyrido[2,1-f][1,2,4]triazin]-7'-yl)oxy)methylmethylcarbonate。根据本发明的一些优选方面,本发明提供了式(1)化合物的a型晶型,其x-射线粉末衍射图(使用辐射源为cu-kα,下同)在2θ角为3.10°±0.2°、8.74°±0.2°、15.44°±0.2°、21.91°±0.2°处有特征峰。进一步地,所述式(1)化合物的a型晶型的x-射线粉末衍射图还在2θ角为13.08°±0.2°、26.35°±0.2°和30.83°±0.2°中的一处或多处具有特征峰。根据本发明的一些具体方面,所述式(1)化合物的a型晶型以热重分析法测定的图谱中显示加热到26.8±2℃时开始失重,加热至150±2℃时失重1.8±0.2%。根据本发明的一些具体方面,所述式(1)化合物的a型晶型以差示扫描量热法测定的图谱中显示有一个吸热峰,显示a型晶型的熔点起始温度为230.5±2℃,在232±2℃时具有吸热峰。根据本发明的一个具体且优选的方面,所述式(1)化合物的a型晶型为无水晶型。根据本发明的一些优选方面,本发明提供了式(1)化合物的b型晶型,其x-射线粉末衍射图在2θ角为8.42°±0.2°、14.27°±0.2°、16.04°±0.2°和25.41°±0.2°处有特征峰。进一步地,所述式(1)化合物的b型晶型的x-射线粉末衍射图还在2θ角为10.75°±0.2°、16.87°±0.2°中的一处或两处具有特征峰。进一步地,所述式(1)化合物的b型晶型的x-射线粉末衍射图还在2θ角为18.21°±0.2°、18.78°±0.2°、19.26°±0.2°、19.60°±0.2°、20.40°±0.2°、21.39°±0.2°、21.66°±0.2°、23.38°±0.2°、27.32°±0.2°、29.17°±0.2°和34.08°±0.2°中的一处或多处具有特征峰。根据本发明的一些具体方面,所述式(1)化合物的b型晶型以热重分析法测定的图谱中显示在被加热到22.6±2℃时开始失重,加热至150±2℃时失重2.2±0.2%。根据本发明的一些具体方面,所述式(1)化合物的b型晶型以差示扫描量热法测定的图谱中显示有两个吸热峰,两个吸热峰的起始温度分别为208.5.5±2℃、233.8±2℃,在213.5±2℃、235.1±2℃时分别具有吸热峰。根据本发明的一个具体方面,所述式(1)化合物的b型晶型为无水晶型。根据本发明的一些优选方面,本发明提供了式(1)化合物的c型晶型,其x-射线粉末衍射图在2θ角为7.73°±0.2°、17.13°±0.2°、20.08°±0.2°、21.74°±0.2°处有特征峰。进一步地,所述式(1)化合物的c型晶型的x-射线粉末衍射图还在2θ角为13.13°±0.2°、13.65°±0.2°、20.98°±0.2°和23.22°±0.2°中的一处或多处具有特征峰。进一步地,所述式(1)化合物的c型晶型的x-射线粉末衍射图还在2θ角为12.51°±0.2°、14.76°±0.2°、15.21°±0.2°、18.39°±0.2°和24.13°±0.2°中的一处或多处具有特征峰。根据本发明的一些具体方面,所述式(1)化合物的c型晶型以热重分析法测定的图谱中显示在被加热到25.9±2℃时开始失重,加热至150±2℃时失重12.1±0.2%。根据本发明的一些具体方面,所述式(1)化合物的c型晶型以差示扫描量热法测定的图谱中显示有两个吸热峰,两个吸热峰的起始温度分别为86.8±2℃、233.9±2℃,在94.9±2℃、234.8±2℃时分别具有吸热峰。根据本发明的一个具体方面,所述式(1)化合物的c型晶型为式(1)化合物的四氢呋喃溶剂合物。根据本发明的一些优选方面,本发明提供了式(1)化合物的d型晶型,其x-射线粉末衍射图在2θ角为7.94°±0.2°、22.16°±0.2°、28.03°±0.2°处有特征峰。进一步地,所述式(1)化合物的d型晶型的x-射线粉末衍射图还在2θ角为15.09°±0.2°、15.50°±0.2°、19.63°±0.2°、23.56°±0.2°、25.86°±0.2°中的一处或多处具有特征峰。根据本发明的一些具体方面,所述式(1)化合物的d型晶型以热重分析法测定的图谱中显示在被加热到23.5±2℃时开始失重,加热至150±2℃时失重7.5±0.2%。根据本发明的一些具体方面,所述式(1)化合物的d型晶型以差示扫描量热法测定的图谱中显示有两个吸热峰,两个吸热峰的起始温度分别为113.2±2℃、230.6±2℃,在140.4±2℃、232.4±2℃时分别具有吸热峰。根据本发明的一个具体方面,所述式(1)化合物的d型晶型为式(1)化合物的n-甲基吡咯烷酮溶剂合物。根据本发明的一些优选方面,本发明提供了式(1)化合物的e型晶型,其x-射线粉末衍射图在2θ角为8.01°±0.2°、8.78°±0.2°、26.33°±0.2°处有特征峰。进一步地,所述式(1)化合物的e型晶型的x-射线粉末衍射图还在2θ角为4.44°±0.2°、17.56°±0.2°、21.95°±0.2°、22.25°±0.2°中的一处或多处具有特征峰。进一步地,所述式(1)化合物的e型晶型的x-射线粉末衍射图还在2θ角为5.87°±0.2°、19.64°±0.2°、28.15°±0.2°、29.07°±0.2°、30.86°±0.2°中的一处或多处具有特征峰。根据本发明的一些优选方面,本发明提供了式(1)化合物的f型晶型,其x-射线粉末衍射图在2θ角为5.77°±0.2°、8.69°±0.2°、17.48°±0.2°、22.84°±0.2°处有特征峰。进一步地,所述式(1)化合物的f型晶型的x-射线粉末衍射图还在2θ角为11.61°±0.2°、19.47°±0.2°中的一处或两处具有特征峰。进一步地,所述式(1)化合物的f型晶型的x-射线粉末衍射图还在2θ角为11.98°±0.2°、13.84°±0.2°、14.30°±0.2°、18.33°±0.2°、18.95°±0.2°中的一处或多处具有特征峰。根据本发明的一些具体方面,所述式(1)化合物的f型晶型以热重分析法测定的图谱中显示在被加热到29.6±2℃时开始失重,加热至150±2℃时失重1.2±0.2%。根据本发明的一些具体方面,所述式(1)化合物的f型晶型以差示扫描量热法测定的图谱中显示有一个吸热峰,吸热峰的起始温度为231.1±2℃,在234.5±2℃时具有吸热峰。根据本发明的一个具体方面,所述式(1)化合物的f型晶型为无水晶型。根据本发明的一些优选方面,本发明还提供了式(1)化合物单晶形式的结晶,其属于斜方晶系,空间群p212121,单位晶胞参数为:α=90°±0.2°,β=90°±0.2°,γ=90°±0.2°。根据本发明的一些优选方面,所述单位晶胞参数为:α=89.9-90.1°,β=89.9-90.1°,γ=89.9-90.1°。根据本发明的一些具体且优选的方面,所述单位晶胞参数为:α=89.95-90.05°,β=89.95-90.05°,γ=89.95-90.05°。在一个具体实施方式中,所述的单位晶胞参数中:α=β=γ=90°。根据本发明的一个具体方面,所述单晶的单位晶胞包含8个分子。根据本发明的一些具体且优选的方面,所述单晶的单位晶胞体积为根据本发明的一个具体方面,所述单晶的单位晶胞体积为根据本发明的一些具体且优选的方面,所述单晶的计算密度为1.480±0.1g/cm3。根据本发明的一个具体方面,所述单晶的计算密度为1.480g/cm3。根据本发明的一些优选方面,所述单晶的制备方法包括:将式(1)化合物溶解于醇类溶剂中,过滤,滤液在室温下挥发溶剂,析出晶体,过滤收集所得晶体并于室温晾干,制得。根据本发明的一些具体方面,所述醇类溶剂包括甲醇、乙醇等。本发明同时又提供了一种技术方案:本发明提供了上述所述的式(1)化合物的a型晶型的制备方法,所述方法包括:1)将式(1)化合物加入到酯类溶剂(例如可以是乙酸异丙酯、乙酸乙酯等)、醇类溶剂(例如可以是甲醇、乙醇、异丙醇等)和酮类溶剂(例如可以是丙酮、丁酮等)中的一种或多种的混合体系中溶解,再与烃类溶剂(例如可以是正庚烷等)混合,结晶,过滤,干燥,得到式(1)化合物的a型晶型;或2)将式(1)化合物加入到酮类溶剂(例如可以是丙酮等)中,室温挥发,得固体,对所得固体进行干燥,得到式(1)化合物的a型晶型;或3)将式(1)化合物加入到醚类溶剂(例如可以是甲基叔丁基醚等)中,室温下搅拌,过滤得固体,对所得固体进行干燥,得到式(1)化合物的a型晶型;或3)将式(1)化合物加入到水中,在45-55℃下搅拌,过滤得固体,对所得固体进行干燥,得到式(1)化合物的a型晶型;或4)将式(1)化合物加入烃类溶剂(例如可以是甲苯等)和醚类溶剂(例如可以是甲基叔丁基醚等)的混合溶剂中,在设定温度下搅拌1~3h,然后以0.1±0.05℃/min的速率进行升温或降温,使体系的温度在所述设定温度至5℃之间循环多次,最终置于3-7℃下搅拌,过滤得到固体,对所得固体进行干燥,得到式(1)化合物的a型晶型,所述设定温度为45~55℃。本发明同时又提供了一种技术方案:本发明提供了上述所述的式(1)化合物的b型晶型的制备方法,所述方法包括:1)将式(1)化合物加入到醇类溶剂(例如可以是甲醇等)或酮类溶剂(例如可以是丙酮等)或其混合体系中溶解,再与水混合,结晶,过滤得固体,对所得固体进行干燥,得到式(1)化合物的b型晶型;或2)将式(1)化合物加入到卤代烃类溶剂(例如可以是氯仿等)中,室温挥发,得固体,对所得固体进行干燥,得到式(1)化合物的b型晶型;或3)将式(1)化合物加入到烃类溶剂(例如可以是甲苯等)中,溶解室温下搅拌,过滤得固体,对所得固体进行干燥,得到式(1)化合物的b型晶型;或4)将式(1)化合物加入到2-甲基四氢呋喃中,置于第一设定温度下搅拌,过滤取上清液,将所得上清液以0.1±0.05℃/min的速率从所述第一设定温度降温至第二设定温度后并在第二设定温度保持恒温,收集析出的固体,对所得固体进行干燥,得到式(1)化合物的b型晶型,所述第一设定温度为45~55℃,所述第二设定温度为0~10℃。本发明同时又提供了一种技术方案:本发明提供了上述所述的式(1)化合物的c型晶型的制备方法:将式(1)化合物加入到四氢呋喃和烃类溶剂的混合体系中形成浑浊液,在第一设定温度下搅拌,过滤取上清液,将所得上清液以0.1±0.05℃/min的速率从第一设定温度降温至第二设定温度后并在第二设定温度保持恒温,收集析出的固体,对所得固体进行干燥,得到式(1)化合物的c型晶型,所述第一设定温度为45~55℃,所述第二设定温度为0~10℃。本发明同时又提供了一种技术方案:本发明提供了上述所述的式(1)化合物的d型晶型的制备方法:将式(1)化合物加入到n-甲基吡咯烷酮和水的混合体系中形成浑浊液,将所得浑浊液置于45-55℃下搅拌,过滤得固体,对所得固体进行干燥,得到式(1)化合物的d型晶型。本发明同时又提供了一种技术方案:本发明提供了上述所述的式(1)化合物的e型晶型的制备方法:所述方法包括:将式(1)化合物溶解在n,n-二甲基乙酰胺中,在水或醇类溶剂的氛围中通过气液扩散得到固体,过滤得式(1)化合物的e型晶型。本发明同时又提供了一种技术方案:本发明提供了上述所述的式(1)化合物的f型晶型的制备方法,所述方法包括:将式(1)化合物加入到酯类溶剂、醇类溶剂或其混合体系中形成浑浊液,在0-10℃搅拌,过滤得固体,对所得固体进行干燥,得到式(1)化合物的f型晶型。根据本发明的一些具体且优选的方面,所述酯类溶剂可以为乙酸乙酯。根据本发明的一些具体且优选的方面,所述醇类溶剂可以为甲醇和/或乙醇。根据本发明的一些具体且优选的方面,所述搅拌的时间为2-4h。本发明同时又提供了一种技术方案:本发明提供了一种药物组合物,其含有上述所述的结晶及药学上可接受的载体。本发明同时又提供了一种技术方案:本发明提供了上述所述的结晶在用于制备抗流感病毒药物中的应用。为了帮助理解本申请公开的各种实施方案,提供以下说明:“式(1)化合物的溶剂合物”,是式(1)化合物与溶剂通过氢键、盐键相互作用而形成的物质。“任意形态的式(1)化合物”,包含式(1)化合物的无定型、晶型、溶剂合物等任意存在的形式。“异构体”是指具有相同分子式但是在其原子的键合性质或键合顺序或其原子空间排列方面有区别的化合物。在其原子空间排列方面不同的异构体被称作“立体异构体”。彼此为非镜像的立体异构体被称作“非对映异构体”,彼此互为实物与镜像而不可重叠的立体异构体被称作“对映异构体”。当化合物具有不对称中心时,例如,如果碳原子与四个不同基团键合时,可能存在一对对映体。对映体可以其不对称中心的绝对构型为特征并且通过cahn-ingold-prelog的r和s顺序规则进行描述,或根据其中分子使偏振光平面旋转并被指定为右旋或左旋(即,分别为(+)或(-)异构体)的方式进行描述。手性化合物绝对构型的测定方法,主要有单晶x-射线衍射法、核磁共振法和圆二色谱法。“药学上可接受的载体”指用于治疗剂给药的载体,包括各种赋形剂和稀释剂。该术语指这样一些药剂载体:它们本身并不是必要的活性成分,且施用后没有过分的毒性。合适的载体是本领域普通技术人员所熟知的。在remington'spharmaceuticalsciences(mackpub.co.,n.j.1991)中可找到关于药学上可接受的赋形剂的充分讨论。在组合物中药学上可接受的载体可包括液体,如水、盐水、甘油和乙醇。另外,这些载体中还可存在辅助性的物质,如崩解剂、润湿剂、乳化剂、ph缓冲物质等。x-射线粉末衍射图谱对于特定的晶型具有特征性。判断是否与已知晶型相同时,应该注意的是峰的相对位置(即2θ)而不是它们的相对强度。这是由于谱图的相对强度会因为晶体条件、粒径和其它测定条件的差异产生的优势取向效果而变化,特别是低强度峰值(强度小于20%)在某些情况下可能不存在,衍射峰的相对强度对晶型的确定并非是特征性的。另外,本领域知道,用x射线衍射测定化合物的结晶时,由于测定的仪器或测定的条件等的影响,同一个晶型的2θ值可存在一定的测量误差,约为±0.2°。因此,在确定每种结晶结构时,应该将此误差考虑在内。在xrd图谱中通常用2θ角或晶面距d值表示峰位置,两者之间具有简单的换算关系:d=λ/2sinθ,其中d代表晶面间距d值,λ代表入射x射线的波长,θ为衍射角。还应特别指出的是,在混合物的鉴定中,由于含量下降等因素会造成部分衍射线缺失。dsc测定当晶体由于其晶体结构发生变化或晶体熔融而吸收或释放热时的转变温度。对于同种化合物的同种晶型,在连续的分析中,热转变温度和熔点误差典型的在约5℃之内。当我们说一个化合物具有一给定的dsc峰或熔点时,这是指该dsc峰或熔点±5℃。需要指出的是对于混合物而言,其dsc峰或熔点可能会在更大的范围内变动。此外,由于在物质熔化的过程中伴有分解,因此熔化温度与升温速率相关。由于以上技术方案的实施,本发明与现有技术相比存在如下优势:本发明提供了新型吡啶酮衍生物,该化合物生物利用度高,在人体服用后可转化为对流感病毒a型和流感病毒b型有极强的抑制活性的化合物,可以单独用于临床治疗或和其他抗流感药物例如神经氨酸酶抑制剂、核苷类药物、pb2抑制剂联合用药,在临床上可快速治愈流感病人,这些化合物在活性、药代动力学特性(如生物利用度)以及细胞毒性等中的至少一个方面优于现有的吡啶酮衍生物。同时本发明提供的式(1)化合物或其溶剂合物的结晶在稳定性、引湿性、溶解性、存储性等方面符合药用的要求,适于在制备治疗流感药物中的应用。附图说明图1为实施例3制得式(1)化合物的a型晶型的xrpd谱图;图2为实施例3制得式(1)化合物的a型晶型的tga谱图;图3为实施例3制得式(1)化合物的a型晶型的dsc谱图;图4为实施例4制得式(1)化合物的b型晶型的xrpd谱图;图5为实施例4制得式(1)化合物的b型晶型的tga谱图;图6为实施例4制得式(1)化合物的b型晶型的dsc谱图;图7为实施例5制得式(1)化合物的c型晶型的xrpd谱图;图8为实施例5制得式(1)化合物的c型晶型的tga谱图;图9为实施例5制得式(1)化合物的c型晶型的dsc谱图;图10为实施例6制得式(1)化合物的d型晶型的xrpd谱图;图11为实施例6制得式(1)化合物的d型晶型的tga谱图;图12为实施例6制得式(1)化合物的d型晶型的dsc谱图;图13为实施例7制得式(1)化合物的e型晶型的xrpd谱图;图14为实施例8制得式(1)化合物的f型晶型的xrpd谱图;图15为实施例8制得式(1)化合物的f型晶型的tga谱图;图16为实施例8制得式(1)化合物的f型晶型的dsc谱图;图17为实施例8制得式(1)化合物的f型晶型的单晶的x射线单晶衍射谱图;图18为实施例10制得式(1)化合物的f型晶型的动态水分吸附谱图。具体实施方式实验所用的测试仪器1.单晶x-射线衍射仪器型号:brukerd8venture单晶衍射仪光源:mo靶x射线:mo-k探测器:cmos面探测器分辨率:电流电压:50kv,1.4a2.x-射线粉末衍射(xrpd)仪器型号:panalyticalx'pert3powerx射线:cu,kα,kα11.540598;kα21.544426x射线光管设定:45kv,40ma发散狭缝:1/8°扫描模式:continuous扫描范围(2θ):3°-40°扫描步长(2θ):0.0263°3.差示扫描量热(dsc)仪器型号:taq2000/2500型差示扫描量热仪升温速率:10℃/min温度范围:25℃至设置终点温度保护气体:氮气4.热重分析(tga)仪器型号:taq5000/5500型热重分析仪升温速率:10℃/min温度范围:室温至设置终点温度保护气体:氮气5.动态水分吸附(dvs)仪器型号:smsdvsintrinsic型水分吸附分析仪温度:25℃rh范围:0%rh-95%rh保护气体:氮气下面结合具体实施例,对本发明做进一步的说明:实施例1式(1)化合物的制备路线如下:式(10)化合物的制备:式(11)化合物(388mg,1mmol)溶于3ml二氯甲烷,加入1ml三氟乙酸,室温搅拌3小时。tlc显示反应完全,加3n氢氧化钠溶液调至ph=9-10。二氯甲烷萃取,有机相合并,饱和食盐水洗,干燥后浓缩得270mg固体,直接用于下一步。式(8)化合物的制备:式(9)将化合物(1.0g,7.8mmol)溶于10ml无水四氢呋喃中,-78℃条件下,氮气保护下缓慢滴加2.5m正丁基锂溶液(3.1ml,7.8mmol)。滴完后在此温度继续搅拌反应2小时。然后滴加氯甲酸烯丙酯(0.94g,7.8mmol)。滴毕继续搅拌反应2h,tlc监测原料基本反应完毕,将反应液倒入饱和氯化铵溶液中淬灭,乙酸乙酯(15ml×3)萃取。合并有机相,无水硫酸钠干燥,浓缩蒸干得1.65g油状物。式(7)化合物的制备:将式(8)化合物(1.65g,7.8mmol)溶于15ml无水四氢呋喃中,氮气保护,于-78℃缓慢滴加1m二异丁基氢化铝溶液(11.7ml,11.7mmol)。滴完后在此温度继续搅拌反应2小时。tlc监测原料基本反应完毕,将反应液倒入饱和酒石酸钾钠溶液中淬灭,乙酸乙酯(20ml×3)萃取。合并有机相,无水硫酸钠干燥,浓缩蒸干得1.57g油状物。式(6)化合物的制备:将式(7)化合物(1.57g,7.4mmol)溶于15ml甲醇,加入对甲苯磺酸一水合物(140mg,0.74mmol),于室温搅拌过夜。tlc监测原料基本反应完毕,加入饱和碳酸氢钠溶液调至中性,浓缩。剩余物经柱层析分离得0.86g黄色油状物。式(5)化合物的制备:式(10)化合物(270mg,0.94mmol)和式(6)化合物(255mg,1.13mmol)溶于5ml乙腈。氮气保护,在-20℃下滴加1m四氯化锡的二氯甲烷溶液(1.4ml,1.41mmol)。滴完后在此温度搅拌3小时。加入饱和碳酸氢钠溶液,搅拌30min,分液,水相用二氯甲烷萃取。有机相合并,饱和食盐水洗,干燥后浓缩得428mg粗产品。式(4)化合物的制备:式(5)化合物(428mg,0.89mmol)溶于5ml四氢呋喃,加入四(三苯基膦)钯(104mg,0.09mmol)和吗啉(774mg,8.9mmol),于室温反应2小时。点板反应结束。浓缩,剩余物柱层析得到216mg产品。式(3)化合物的制备:式(4)化合物(216mg,0.61mmol)和式(12)化合物(242mg,0.92mmol)在3mlt3p的乙酸乙酯溶液中于100℃密闭反应3小时。冷却,加饱和nahco3稀释,然后乙酸乙酯萃取。有机相合并,干燥后浓缩,柱层析分离得200mg粗品,然后手性柱分离得到40mg产品。式(2)化合物的制备:式(3)化合物(40mg,0.067mmol)和氯化锂(20mg,0.48mmol)在1mldma中于100℃反应3小时。反应完全后加10ml水稀释,用2n盐酸调ph至3-4。过滤,固体抽干得25mg产品。1hnmr(400mhz,cdcl3)δ:7.05-7.15(m,5h),6.85(m,1h),6.70(d,1h,j=7.6hz),5.78(d,1h,j=7.6hz),5.3(m,2h),4.69(d,1h,j=6.8hz),4.17(d,1h,j=14hz),4.09(d,1h,j=14hz),3.90(m,1h),3.69(m,1h),3.44(d,1h,j=15.2hz),0.95(m,1h),0.74(m,3h);esi-msm/z(m+h)+510.1。式(1)化合物的制备:式(2)化合物(40mg,0.08mmol),氯甲基甲基碳酸酯(25mg,0.2mmol),碳酸钾(28mg,0.2mmol)和碘化钾(3mg,0.02mmol)在1mln,n-二甲基乙酰胺中于60℃反应5小时。点板反应完全,加水淬灭反应,然后用1n稀盐酸调ph至3-4。固体过滤后干燥,柱层析得35mg产品,即为式(1)化合物,该固体经分析为无定形,用作后续实施例中制备各种晶型的起始原料。1hnmr(400mhz,dmso-d6)δ:7.40-7.42(m,2h),7.25(d,1h,j=7.6hz),7.15(m,1h),7.10(m,1h),7.00(d,1h,j=7.2hz),6.84(t,1h,j=7.6hz),5.75(m,4h),5.43(d,1h,j=16.4hz),4.57(dd,1h,j=3.2,9.6hz),3.96-4.03(m,3h),3.73(s,3h),3.51(t,1h,j=10.0hz),3.41(s,1h),0.75(t,2h,j=8.4hz),0.50(m,2h);esi-msm/z(m+h)+598.1。实施例2式(1)化合物的药效实验2.1、体外生物活性研究和细胞毒性研究实际用药中,式(1)化合物为式(2)化合物的前药,其在体内转化为活性药物(式(2)化合物)而发挥药效,式(2)化合物的活性数据以及毒性数据参见表a。体外生物活性研究的试验方法:将mdck细胞以2,000细胞每孔的密度种入384孔细胞培养板中,随后置于37℃,5%co2培养箱中培养过夜。第二天,将式(2)化合物稀释后分别加入到细胞孔内(3倍倍比稀释,8个测试浓度点),流感病毒a/pr/8/34(h1n1)株随后以每孔2*tcid90加入细胞培养孔中,培养基中dmso终浓度为0.5%。细胞板置于37℃,5%co2培养箱中培养5天。培养5天后使用细胞活力检测试剂盒cck8检测细胞活性。原始数据用graphpadprism软件对化合物的抑制率和细胞毒性进行非线性拟合分析,得到ec50值(结果参见表a)。细胞毒性研究的研究方法:式(2)化合物的细胞毒性测定和抗病毒活性测定平行进行,除了不加病毒,其它的实验条件和抗病毒活性实验一致。培养5天后使用细胞活力检测试剂盒cck8检测细胞活性。原始数据用于化合物细胞毒性(cc50)计算(结果参见表a)。表a.化合物对于流感病毒a/pr/8/34(h1n1)的抑制活性以及毒性结果表明,根据本发明的化合物相比对照化合物,具有更优秀的抑制h1n1的活性,并且具有很低的细胞毒性。2.2、大鼠pk研究静脉注射:准确称量式(2)化合物供试品约2mg,加入适量dma,涡旋振荡使固体物质完全溶解;再加入适量体积的30%solutolhs-15水溶液,涡旋振荡后再加入saline,使得dma:30%solutolhs-15:saline=20:20:60(v/v/v),涡旋振荡使液体混合均匀,并过滤,得浓度为0.05mg·ml-1的给药制剂。sd大鼠单次静脉注射给予0.25mg·kg-1的式(2)化合物静注给药制剂。分别于给药前及给药后0.083、0.25、0.5、1、2、4、8、12和24h,由颈静脉采血0.20ml,置于edta-k2抗凝管中。立即准确吸取150μl全血,加到已加入450μl乙腈的试管中蛋白沉淀,涡旋振荡,置于湿冰上。保存在-90~-60℃冰箱,用于生物样品分析。利用lc-ms/ms分析方法,测定s-d大鼠血浆中对应化合物的浓度。采用pharsightphoenix7.0中的非房室模型计算相应的药代动力学参数。结果参见表b。灌胃给药:准确称量式(1)化合物供试品4mg,加入适量peg400,涡旋振荡使固体物质溶解;再加入适量体积的30%solutolhs-15水溶液和saline,使得peg400:30%solutolhs15(w/v):saline=2:2:6(v/v/v),漩涡振荡,得浓度为0.3mg·ml-1的给药制剂。sd大鼠单次灌胃给予3.0mg·kg-1的式(1)化合物口服给药制剂,然后于给药前及给药后0.25、0.5、1、2、4、8、12和24h测定s-d大鼠血浆中对应式(2)化合物的浓度。结果参见表c。表b.式(2)化合物的pk参数(静脉注射)表c.式(1)化合物的pk参数(灌胃)以上结果表明本发明的式(1)化合物体内清除率低,具有较长的半衰期,生物利用度高,在体内有较高的吸收。2.3、小鼠药效雌性balb/c小鼠通过滴鼻方式接种甲型流感病毒(h1n1,a/wsn/33)建立iav小鼠感染模型。每天2次口服溶媒、式(1)化合物(15mpk)或磷酸奥司他韦(15mpk)。试验期间每天监测动物体重及存活状态,并在第5天处死部分动物取肺组织用于病毒滴度检测,剩余小鼠用于存活率监测。通过肺组织病毒滴度,小鼠体重变化及存活率确定测试化合物的体内抗流感病毒药效。肺组织病毒滴度:病毒感染后第5天,溶媒组小鼠肺组织中病毒滴度平均值达7.20log10(每克肺组织空斑数),磷酸奥司他韦组小鼠肺组织中病毒滴度平均值为3.74log10(每克肺组织空斑数)。与溶媒组相比,磷酸奥司他韦显著抑制了病毒在小鼠体内的复制,病毒的滴度平均值降低3.46log10(每克肺组织空斑数),结果具有非常显著性统计学差异(p<0.01),显示出预期的药效;感染小鼠经受试式(1)化合物治疗后,第5天小鼠肺组织中病毒滴度平均值为3.28log10(每克肺组织空斑数),与溶媒组相比,受试化合物显著抑制了病毒在小鼠体内的复制,病毒滴度平均值降低3.92log10(每克肺组织空斑数),结果具有极其显著性统计学差异(p<0.001),并优于对照化合物磷酸奥司他韦(表d)。表d.肺组织病毒滴度**p<0.01表示具有非常显著性差异,***p<0.001表示具有极其显著性差异体重变化及结果分析:溶媒组小鼠在感染后第3天开始出现显著的体重下降,随后持续下降甚至死亡;磷酸奥司他韦组和式(1)化合物组小鼠在实验过程中体重维持稳定,未见明显下降,小鼠健康状况良好。生存率及结果分析:溶媒组小鼠在感染后在第7天发现死亡,至第10天小鼠全部死亡或因体重下降至人道终点而被安乐死,存活率为0%;磷酸奥司他韦和式(1)化合物组小鼠在实验过程中维持健康,所有动物均存活至预定实验终点,存活率为100%,显示了极佳的体内抗流感药效。实施例3:式(1)化合物的a型晶型的制备方法1:称取式(1)化合物15毫克,用1毫升乙酸异丙酯完全溶解。向该澄清溶液中边搅拌边缓慢加入4毫升正庚烷,有固体析出。搅拌1小时后过滤,固体在50度鼓风干燥4h,得13.6毫克,收率90.7%,hplc纯度99.3%。方法2:称取式(1)化合物15毫克,用1毫升乙酸乙酯完全溶解。在20毫升小瓶中加入4毫升正庚烷,将化合物的乙酸乙酯溶液缓慢加入到正庚烷中,边搅拌边滴加,有固体析出。搅拌1小时后过滤,固体在50度鼓风干燥4h,得14.0毫克,收率93.3%,hplc纯度99.1%。方法3:称取式(1)化合物15毫克,用1毫升丙酮完全溶解,然后在室温缓慢挥发,收集所得固体,于50度鼓风干燥4h。方法4:称取式(1)化合物15毫克,加入0.5毫升甲基叔丁基醚,得到的浑浊液置于室温下磁力搅拌3天,离心收集所得固体,于50度鼓风干燥4h。方法5:称取式(1)化合物15毫克,加入0.5毫升水,得到的浑浊液置于50℃下磁力搅拌3天,离心收集所得固体,于50度鼓风干燥4h。方法6:称取式(1)化合物15毫克,加入0.1毫升甲苯和0.4毫升甲基叔丁基醚,得到的浑浊液置于50℃下搅拌约2小时后,将样品放置在生化培养箱中,以0.1℃/分钟的速率升降温,并进行50-5℃三次循环升降温测试。样品最终被置于5℃搅拌,离心收集所得固体,于50度鼓风干燥4h。对方法1得到的固体进行xrpd测试,谱图如图1所示,在衍射角2θ=3.10、8.74、13.08、15.44、21.91、26.35、30.83度处有特征峰,2θ误差范围为±0.2度。其x-射线粉末衍射数据如表1所示。tga(图2)和dsc结果(图3)显示样品加热至150℃时有1.8%的失重,且在232.0℃(峰值)有一个吸热峰,式(1)化合物a型晶型晶型为无水晶型。表1:式(1)化合物的a型晶型的xrpd图谱详情对其它方法得到的固体分别进行了xrpd测试,测试谱图均与图1基本一致,表明所得固体为式(1)化合物的a型晶型。实施例4:式(1)化合物的b型晶型的制备方法1:称取式(1)化合物15毫克,用1毫升甲醇完全溶解。向该澄清溶液中边搅拌边缓慢加入4毫升水,有固体析出。搅拌1小时后过滤,固体在50度鼓风干燥4h,得14.1毫克,收率94%,hplc纯度99.2%。方法2:称取式(1)化合物15毫克,用1毫升丙酮完全溶解。在20毫升小瓶中加入4毫升水,将化合物的丙酮溶液缓慢加入到水中,边搅拌边滴加,有固体析出。搅拌1小时后过滤,固体在50度鼓风干燥4h,得13.8毫克,收率92%,hplc纯度99.2%。方法3:称取式(1)化合物15毫克,用1毫升氯仿完全溶解,然后在室温缓慢挥发,收集所得固体,于50度鼓风干燥4h。方法4:称取式(1)化合物15毫克,加入0.5毫升甲苯,得到的浑浊液置于室温下磁力搅拌3天,离心收集所得固体,于50度鼓风干燥4h。方法5:称取式(1)化合物15毫克,加入0.5毫升2-甲基四氢呋喃,得到的浑浊液置于50℃下搅拌2小时后,过滤取上清液。将所得上清液以0.1℃/分钟从50℃降温至5℃后并在5℃保持恒温。收集析出的固体,于50度鼓风干燥4h。对方法1得到的固体进行xrpd测试,谱图如图4所示,在衍射角2θ=8.42、10.75、14.27、16.04、16.87、19.60、20.40、21.39、21.66、23.38、25.41、27.32、29.17、34.08度处有特征峰,2θ误差范围为±0.2度。其x-射线粉末衍射数据如表2所示。tga(图5)和dsc结果(图6)显示样品加热至150℃时有2.2%的失重,且在213.5℃和231.5℃(峰值)有2个吸热峰,式(1)化合物的b型晶型为无水晶型。表2:式(1)化合物的b型晶型的xrpd图谱详情对其它方法得到的固体分别进行了xrpd测试,测试谱图均与图4基本一致,表明所得固体为式(1)化合物的b型晶型。实施例5:式(1)化合物的c型晶型的制备方法:称取式(1)化合物15毫克,加入0.25毫升四氢呋喃和0.25毫升正庚烷,得到的浑浊液置于50℃下搅拌2小时后,过滤取上清液。将所得上清液以0.1℃/分钟从50℃降温至5℃后并在5℃保持恒温。收集析出的固体,于50度鼓风干燥4h。对上述方法得到的固体进行xrpd测试,谱图如图7所示,在衍射角2θ=7.73、12.51、13.13、13.65、14.76、15.21、17.13、18.39、20.08、20.98、21.74、23.22、24.13度处有特征峰,2θ误差范围为±0.2度。其x-射线粉末衍射数据如表3所示。tga(图8)和dsc(图9)结果显示样品加热至150℃时有12.1%的失重,且在94.9℃和234.8℃(峰值)有2个吸热峰,式(1)化合物的c型晶型为thf溶剂合物。表3:式(1)化合物的c型晶型的xrpd图谱详情实施例6:式(1)化合物的d型晶型的制备方法:称取式(1)化合物15毫克,加入0.05毫升n-甲基吡咯烷酮和0.45毫升水,得到的浑浊液置于50℃下搅拌3天后,离心收集固体,并于50度鼓风干燥4h。对上述方法得到的固体进行xrpd测试,谱图如图10所示,在衍射角2θ=7.94、15.09、15.50、19.63、22.16、23.56、25.86、28.03度处有特征峰,2θ误差范围为±0.2度。其x-射线粉末衍射数据如表4所示。tga(图11)和dsc(图12)结果显示样品加热至150℃时有7.5%的失重,且在140.4℃和232.4℃(峰值)有2个吸热峰,式(1)化合物的d型晶型为n-甲基吡咯烷酮溶剂合物。表4:式(1)化合物的d型晶型的xrpd图谱详情实施例7:式(1)化合物的e型晶型的制备方法:称取式(1)化合物15毫克,加入0.5毫升n,n-二甲基乙酰胺溶解,将装有此溶液的3ml小瓶敞口置于装有4ml水的20ml小瓶中,密封20ml小瓶并于室温下静置7天,得到14.3毫克白色固体,收率95.2%,hplc纯度99.2%对上述方法得到的固体进行xrpd测试,谱图如图13所示,在衍射角2θ=4.44、5.87、8.01、8.78、17.56、19.64、21.95、22.25、26.33、28.15、29.07、30.86度处有特征峰,2θ误差范围为±0.2度。其x-射线粉末衍射数据如表5所示。表5:式(1)化合物的e型晶型的xrpd图谱详情实施例8:式(1)化合物的f型晶型的制备方法1:称取式(1)化合物33克,加入40毫升甲醇,在0-10℃搅拌3h后过滤。固体在50度鼓风干燥8h,得31.88克白色固体,收率96.6%,hplc纯度99.4%。方法2:称取式(1)化合物33克,加入40毫升乙醇,在0-10℃搅拌3h后过滤。固体在50度鼓风干燥8h,得30.76克白色固体,收率93.2%,hplc纯度99.5%。方法3:称取式(1)化合物33克,加入40毫升乙酸乙酯,在0-10℃搅拌3h后过滤。固体在50度鼓风干燥8h,得31.24克白色固体,收率94.7%,hplc纯度99.5%。对方法1所得固体进行xrpd测试,谱图如图14所示,在衍射角2θ=5.77、8.69、11.61、11.98、13.84、14.30、17.48、18.33、18.95、19.47、22.84度处有特征峰,2θ误差范围为±0.2度。其x-射线粉末衍射数据如表6所示。tga(图15)和dsc(图16)结果显示样品加热至150℃时有1.2%的失重,且在234.5℃(峰值)有1个吸热峰,式(1)化合物的f型晶型为无水晶型。表6:式(1)化合物的f型晶型的xrpd图谱详情对其它方法得到的固体分别进行了xrpd测试,测试谱图均与图14基本一致,表明所得固体为式(1)化合物的f型晶型。其中,式(1)化合物的f型晶型的单晶通过如下方法制备:将式(1)化合物溶解于甲醇中,过滤,滤液在室温下缓慢挥发甲醇,析出晶体,过滤收集所得晶体并于室温晾干,制得。对所得单晶进行单晶x-射线衍射数据收集并解析其单晶结构。式(1)化合物的单晶结构数据参见表7。单晶结构解析确定了式(1)化合物手性中心的绝对构型。如图17示,根据cahn-ingold-prelog的r和s顺序规则,c15的手性是r,c16的手性是s。表7:式(1)化合物的f型晶型的单晶结构数据实施例9:晶型固态稳定性评估将式(1)化合物的f型晶型样品分别在60℃放置30天,25℃/92.5%rh放置30天,以及接受光照考察15天。通过xrpd和hplc检测样品的物理和化学稳定性。结果表明在各稳定性测试条件下,式(1)化合物的f型晶型样品均未发生晶型转变和hplc纯度降低,表明其在测试条件下具有良好的物理化学稳定性(表8)。表8:式(1)化合物的f型晶型的固态稳定性条件纯度(面积%)纯度/起始(%)晶型变化外观性状起始98.6-否白色粉末60℃/30天98.7100.1否白色粉末25℃/92.5%rh/30天99.0100.4否白色粉末光照*/15天98.599.9否白色粉末*白光:4500lux,紫外光:90μw/cm2实施例10:晶型引湿性评估通过25℃下的动态水分吸附试验(dvs)测量了式(1)化合物的f型晶型样品的引湿性,结果显示在25℃/80%rh条件下样品的吸湿增重为0.10%(图18),表明样品几乎无引湿性。同时,xrpd结果显示式(1)化合物的f型晶型在dvs测试前后晶型未发生转变。对其他晶型的稳定性和引湿性也进行了研究,结果表明,它们均符合药用要求,其中以式(1)化合物的f晶型最为稳定,引湿性最好。以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本
技术领域:
的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1