1,3-二甲基-1H-吡唑-4-甲酸的合成方法与流程
本发明涉及农药中间体领域,尤其涉及1,3-二甲基-1h-吡唑-4-甲酸的合成方法。
背景技术:
吡唑酰胺类化合物作为杀菌剂的开发源于上世纪90年代,日本住友化学和三井化学先后有相关研究开发的报道。进入21世纪,尤其是近几年来,吡唑酰胺类杀菌活性化合物越来越收到人们的关注。农药巨头拜耳、巴斯夫、先正达和杜邦均已进入这一领域的研究开发,各自均有此类杀菌剂新品种处于开发阶段。其特征是新颖的化学结构,广谱高效和可能不同于现有杀菌剂的作用方式。
目前,1,3-二甲基-1h-吡唑-4-甲酸及其衍生物的合成方法主要源于以下三种文献:journalofheterocyclicchemistry,2018,55(4),946-950;
bulletindelasocietechimiquedefrance,1988,3,540-7;
newjournalofchemistry,2019,43(7),3000-3010。
但是上述路线存在着收率低下,环境不友好或者成本较高等不足。
技术实现要素:
本发明克服了现有技术的不足,提供一种是以乙酰乙酸乙酯为原料经缩合关环,甲酰化,还原脱氯,氧化得到作为杀线虫剂中间体的1,3-二甲基-1h-吡唑-4-甲酸的合成方法。
为达到上述目的,本发明采用的技术方案为:提供了1,3-二甲基-1h-吡唑-4-甲酸的合成方法,其特征在于:包括以下步骤:
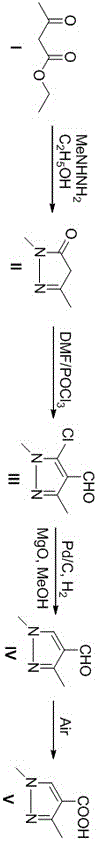
(1)乙酰乙酸乙酯与甲基肼进行缩合关环反应生成1,3-二甲基-5-吡唑酮;
(2)1,3-二甲基-5-吡唑酮与三氯氧磷以及n,n-二甲基甲酰胺反应得到5-氯-1,3-二甲基-1h-吡唑-4-甲醛;
(3)5-氯-1,3-二甲基-1h-吡唑-4-甲醛还原脱氯得到1,3-二甲基-1h-吡唑-4-甲醛;
(4)1,3-二甲基-1h-吡唑-4-甲醛经氧化得到1,3-二甲基-1h-吡唑-4-甲酸。
作为一种优选方案,具体包括以下步骤:
(1)将乙酰乙酸乙酯溶于第一溶剂中;之后向体系中加入甲基肼;加入完毕后升温,乙酰乙酸乙酯与甲基肼缩合关环生成1,3-二甲基-5-吡唑酮;
(2)将三氯氧磷加入n,n-二甲基甲酰胺中;之后向体系中加入1,3-二甲基-5-吡唑酮;升温,反应生成5-氯-1,3-二甲基-1h-吡唑-4-甲醛;
(3)将5-氯-1,3-二甲基-1h-吡唑-4-甲醛溶于第二溶剂中;之后向体系加入pd/c和mgo;之后在氢气条件下通过还原脱氯得到1,3-二甲基-1h-吡唑-4-甲醛;
(4)将1,3-二甲基-1h-吡唑-4-甲醛溶于第三溶剂中;之后在异氰存在的条件下,在空气中通过氧化反应得到1,3-二甲基-1h-吡唑-4-甲酸;
作为一种更优选方案,所述步骤(1)中,甲基肼的加入温度为0-5℃;反应温度为78℃;乙酰乙酸乙酯与甲基肼的摩尔比为1:1-3。
作为一种更优选方案,所述步骤(1)中,第一溶剂为醇类溶剂、二氯甲烷、1,4-二氧六环、乙腈、四氢呋喃、水中的一种或几种。
作为一种更优选方案,所述步骤(2)中,三氯氧磷的加入温度为0-5℃;反应温度为80-100℃。
作为一种更优选方案,所述步骤(2)中,1,3-二甲基-5-吡唑酮、三氯氧磷与n,n-二甲基甲酰胺的重量比为1:1-10:1-10。
作为一种更优选方案,所述步骤(3)中,还原反应温度为20-30℃;5-氯-1,3-二甲基-1h-吡唑-4-甲醛、pd/c与mgo的重量比为1:0.01-0.5:0.01-0.5。
作为一种更优选方案,所述步骤(3)中,第二溶剂为醇类溶剂、n,n-二甲基甲酰胺、二氯甲烷、四氢呋喃、乙腈、二氧六环中的一种或几种。
作为一种更优选方案,所述步骤(4)中,氧化反应温度为10-70℃;异氰为异氰乙酸乙酯、异氰正丁烷、异氰叔丁烷中的一种;1,3-二甲基-1h-吡唑-4-甲醛与异氰的摩尔比为1:0.1-2。
作为一种更优选方案,所述步骤(4)中,第三溶剂为水、醇类溶剂、乙腈、乙酸乙酯、二氯甲烷、n,n-二甲基甲酰胺、四氢呋喃中的一种或几种。
本发明与现有的技术相比有益技术效果在于:提供了作为杀线虫剂中间体的1,3-二甲基-1h-吡唑-4-甲酸的合成方法;在已报道的方法中,通常使用昂贵且高污染性的锰试剂,铬试剂等重金属催化剂,不仅会使反应成本大大提高,而且产生的废料会造成环境污染。而本方法以廉价易得的空气作为氧化源,并成功地以较高收率得到目标产品。故本方法反应成本更低,收率更高,具有更好的经济效益;且避免了重金属氧化所带来的成本及环境污染等问题,为工业化创造了条件。
附图说明
下面结合附图和实施例对本发明进一步说明。
图1为实施例1的合成路线图。
具体实施方式
下面结合具体实施例对本发明作进一步描述。以下实施例仅用于更加清楚地说明本发明的技术方案,而不能以此来限制本发明的保护范围。
实施例1
(1)0℃的冰浴下,将8.8g甲基肼(0.19mol)滴加至乙酰乙酸乙酯(10.0g,0.077mol)的乙醇溶液(20ml)中;滴加完毕,升温至78℃回流,反应2小时;反应结束后,浓缩反应液,再用无水乙醇带水,得9.1g的1,3-二甲基-5-吡唑酮浅黄色固体;纯度95%,收率100%。
(2)0℃的冰浴下,将54ml三氯氧磷滴加至的18ml的n,n-二甲基甲酰胺(dmf)溶液中;滴加完毕后,自然升至室温,加入9.1g的1,3-二甲基-5-吡唑酮(0.08mol);加入完毕后,升温至90℃反应1小时;反应结束后,反应液降至室温并倒至冰水中;之后经过乙酸乙酯萃取,有机相干燥,浓缩等步骤得到8.5g的5-氯-1,3-二甲基-1h-吡唑-4-甲醛浅黄色固体;纯度92%,收率70%。
(3)将8.5g的5-氯-1,3-二甲基-1h-吡唑-4-甲醛(0.054mol)溶于甲醇中,加入pd/c(0.85g,0.1w/w)和mgo(1.7g,0.2w/w);体系用氢气置换三次;在1个大气压氢气的条件下,25℃下搅拌反应16小时;反应完全后,过滤除去固体(滤饼洗涤),滤液减压浓缩,加水溶解,并用乙酸乙酯萃取,有机相浓缩得到6.3g的1,3-二甲基-1h-吡唑-4-甲醛浅黄色固体;纯度93%,收率96%。
(4)将6.3g的1,3-二甲基-1h-吡唑-4-甲醛(0.052mol)和2.9g的异氰乙酸乙酯(0.026mol)加入30ml水中,并在空气中加热至40℃搅拌反应3小时;反应结束后,向反应液加入50%氢氧化钠水溶液混合均匀,之后用二氯甲烷萃取,除去杂质;水相调酸至ph=2-3,过滤,滤饼洗涤,收集固体,得到4.9g的1,3-二甲基-1h-吡唑-4-甲酸;纯度99%,收率79%。
所得1,3-二甲基-1h-吡唑-4-甲酸采用核磁共振检测,结果如下:
1hnmr(dmso-d6):7.91(s,1h),3.82(s,3h),2.31(s,3h)。
结果表明:1,3-二甲基-1h-吡唑-4-甲酸被很好地合成出。
实施例2
本实施例与实施例1相比,仅步骤(1)的反应参数有所改变,其余步骤相同。
步骤(1)具体如下:0℃的冰浴下,将8.7g甲基肼(0.19mol)滴加至乙酰乙酸乙酯(10.0g,0.077mol)的四氢呋喃溶液(20ml)中;滴加完毕,升温至78℃回流,反应3小时;反应结束后,浓缩反应液,再用无水乙醇带水,得7.2g的1,3-二甲基-5-吡唑酮浅黄色固体;纯度96%,收率96%。
实施例3
本实施例与实施例1相比,仅步骤(1)的反应参数有所改变,其余步骤相同。
步骤(1)具体如下:5℃下,将4.4g甲基肼(0.08mol)滴加至乙酰乙酸乙酯(10.0g,0.077mol)的乙醇溶液(20ml)中;滴加完毕,升温至78℃回流,反应8小时;反应结束后,浓缩反应液,再用无水乙醇带水,得4.3g的1,3-二甲基-5-吡唑酮浅黄色固体;纯度94%,收率51%。
实施例4
本实施例与实施例1相比,仅步骤(2)的反应参数有所改变,其余步骤相同。
步骤(2)具体如下:5℃下,将19ml三氯氧磷滴加至的18ml的n,n-二甲基甲酰胺(dmf)溶液中;滴加完毕后,自然升至室温,加入9.1g的1,3-二甲基-5-吡唑酮(0.08mol);加入完毕后,升温至100℃反应1小时;反应结束后,反应液降至室温并倒至冰水中;之后经过乙酸乙酯萃取,有机相干燥,浓缩等步骤得到8.2g的5-氯-1,3-二甲基-1h-吡唑-4-甲醛浅黄色固体;纯度92%,收率65%。
实施例5
本实施例与实施例1相比,仅步骤(3)的反应参数有所改变,其余步骤相同。
步骤(3)具体如下:将8.5g的5-氯-1,3-二甲基-1h-吡唑-4-甲醛(0.054mol)溶于甲醇中,加入pd/c(0.85g,0.1w/w)和mgo(0.8g,0.1w/w);体系用氢气置换三次;在1个大气压氢气的条件下,30℃下搅拌反应24小时;反应完全后,过滤除去固体(滤饼洗涤),滤液减压浓缩,加水溶解,并用乙酸乙酯萃取,有机相浓缩,柱层析纯化得到4.3g的1,3-二甲基-1h-吡唑-4-甲醛浅黄色固体;纯度95%,收率65%。
实施例6
本实施例与实施例1相比,仅步骤(3)的反应参数有所改变,其余步骤相同。
步骤(3)具体如下:将8.5g的5-氯-1,3-二甲基-1h-吡唑-4-甲醛(0.054mol)溶于二氧六环中,加入pd/c(0.9g,0.1w/w)和mgo(1.8g,0.2w/w);体系用氢气置换三次;在1个大气压氢气的条件下,20℃下搅拌反应20小时;反应完全后,过滤除去固体(滤饼洗涤),滤液减压浓缩,加水溶解,并用乙酸乙酯萃取,有机相浓缩,柱层析纯化得到5.1g的1,3-二甲基-1h-吡唑-4-甲醛浅黄色固体;纯度95%,收率78%。
实施例7
本实施例与实施例1相比,仅步骤(4)的反应参数有所改变,其余步骤相同。
步骤(4)具体如下:将6.4g的1,3-二甲基-1h-吡唑-4-甲醛(0.054mol)和2.9g的异氰乙酸乙酯(0.028mol)加入30ml水中,并在空气中加热至70℃搅拌反应1小时;反应结束后,向反应液加入50%氢氧化钠水溶液混合均匀,之后用二氯甲烷萃取,除去杂质;水相调酸至ph=2-3,过滤,滤饼洗涤,收集固体,得到4.1g的1,3-二甲基-1h-吡唑-4-甲酸;纯度98%,收率65%。
实施例8
本实施例与实施例1相比,仅步骤(4)的反应参数有所改变,其余步骤相同。
步骤(4)具体如下:将6.4g的1,3-二甲基-1h-吡唑-4-甲醛(0.054mol)和2.3g的异氰正丁烷(0.028mol)加入30ml水中,并在空气中加热至10℃搅拌反应12小时;反应结束后,向反应液加入50%氢氧化钠水溶液混合均匀,之后用二氯甲烷萃取,除去杂质;水相调酸至ph=2-3,过滤,滤饼洗涤,收集固体,得到4.4g的1,3-二甲基-1h-吡唑-4-甲酸;纯度97%,收率71%。
以上依据本发明的理想实施例为启示,通过上述的说明内容,相关人员完全可以在不偏离本项发明技术思想的范围内,进行多样的变更以及修改。本项发明的技术性范围并不局限于说明书上的内容,必须要根据权利要求范围来确定技术性范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 5-(4-氟苯基)-N,N-二甲基-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 制备1-烷基-3-二氟甲基-5-氟-1h-吡唑-4-甲醛和1-烷基-3-二氟甲基-5-氟-1h-吡唑-4 ...的制作方法
- 三氟甲基吡唑基胍F<sub>1</sub>F<sub>0</sub>-ATP酶抑制剂及其治疗用图
- 3,5-二甲基-1h-吡唑-1-甲脒盐酸盐的制备方法
- 取代吡唑基吡唑衍生物及其作为除草剂的用图
- 取代吡唑基吡唑衍生物及其作为除草剂的用图
- 封端异氰酸酯、涂料组合物、粘接剂组合物和物品的制作方法
- 一种1-(3-甲基-1-苯基-1h-吡唑-5-基)哌嗪的制备方法
- 一种3-二氟甲基-1-甲基-1h-吡唑-4-羧酸乙酯的合成方法
- 一种1,3,5-三甲基-4-吡唑甲酸甲酯的合成方法